Tianjin Key Laboratory of Metabolic Diseases, Department of Physiology and Pathophysiology, Tianjin Medical University, Tianjin, China.
The Center for Non-infectious Liver Diseases, Beijing 302 Military Hospital, Beijing, China.
Cell Mol Gastroenterol Hepatol. 2019;7(1):211-231. doi: 10.1016/j.jcmgh.2018.09.011. Epub 2018 Sep 19.
BACKGROUND & AIMS: Nonalcoholic steatohepatitis (NASH) is an increasingly prevalent nonalcoholic fatty liver disease, characterized by inflammatory cell infiltration and hepatocellular damage. Mammalian target of rapamycin complex 1 (mTORC1) has been investigated extensively in the context of cancer, including hepatocellular carcinoma. However, the role of mTORC1 in NASH remains largely unknown.
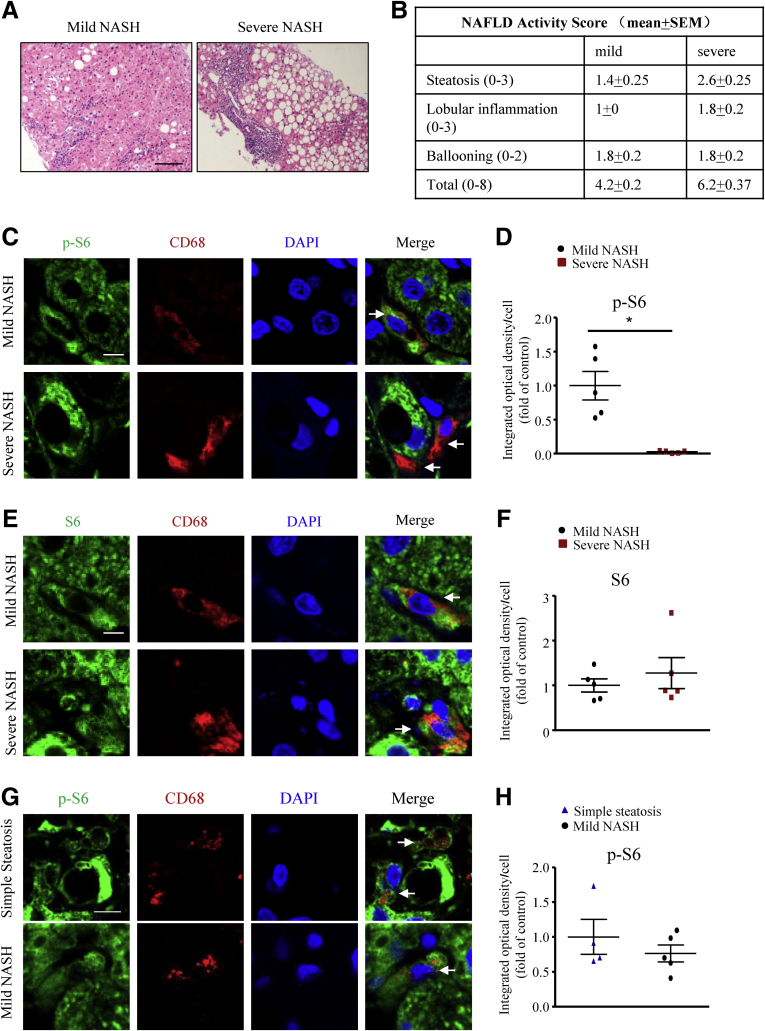
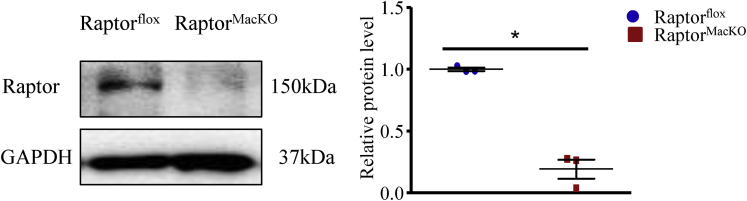
mTORC1 activity in macrophages in human mild and severe NASH liver was compared. Mice with macrophage-specific deletion of the regulatory-associated protein of mTOR (Raptor) subunit and littermate controls were fed a high-fructose, palmitate, and cholesterol diet for 24 weeks or a methionine- and choline-deficient diet for 4 weeks to develop NASH.
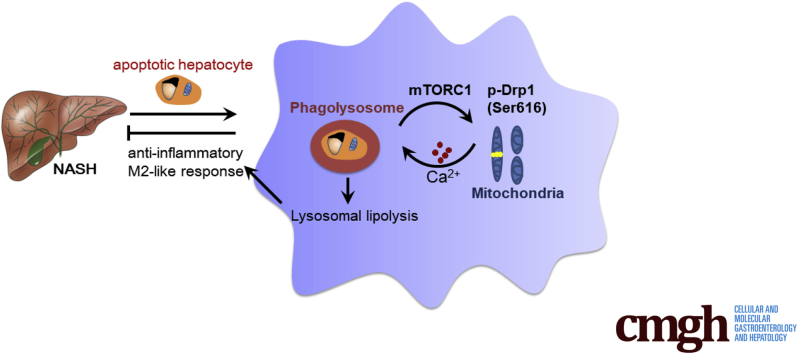
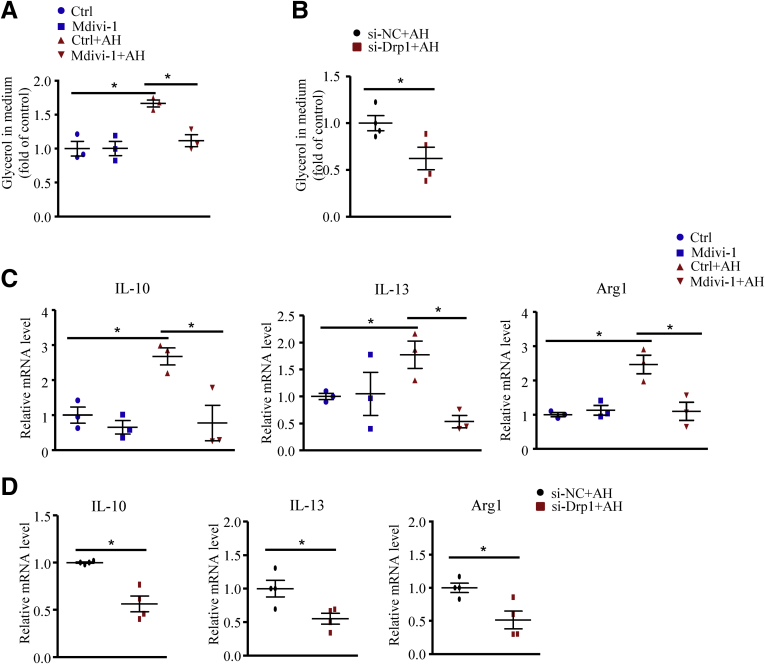
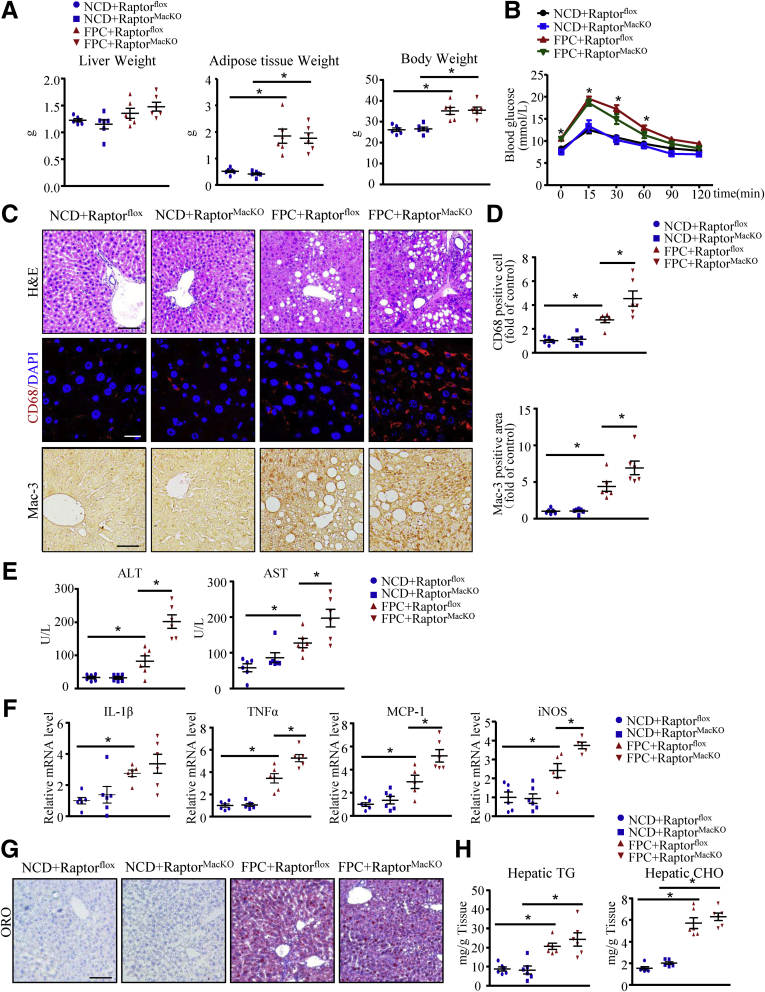
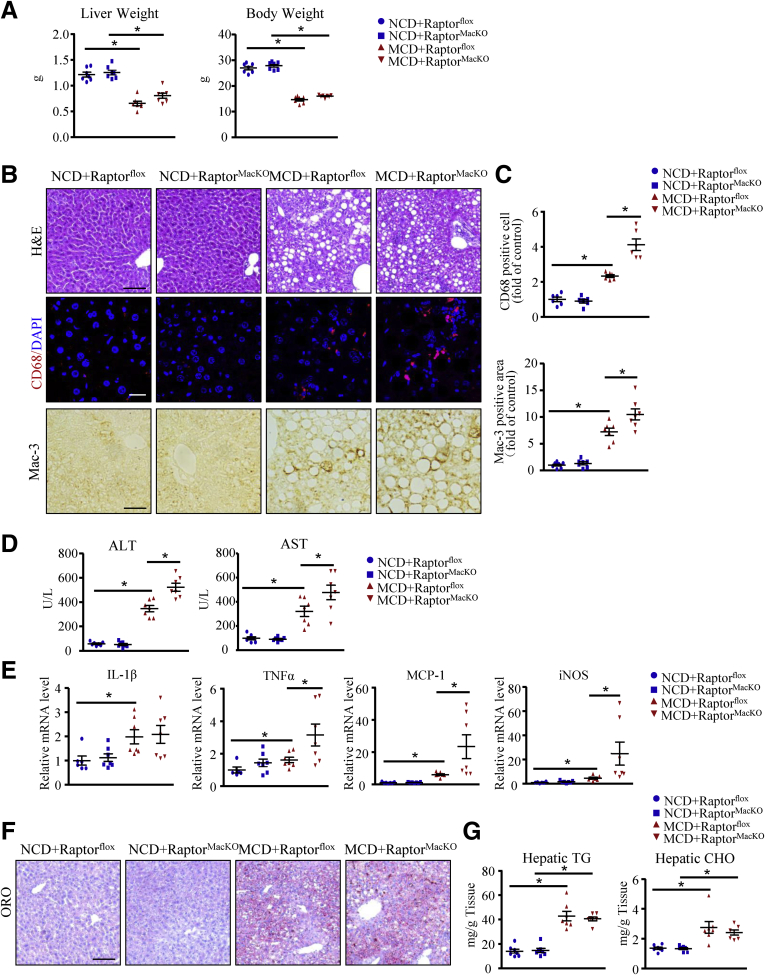
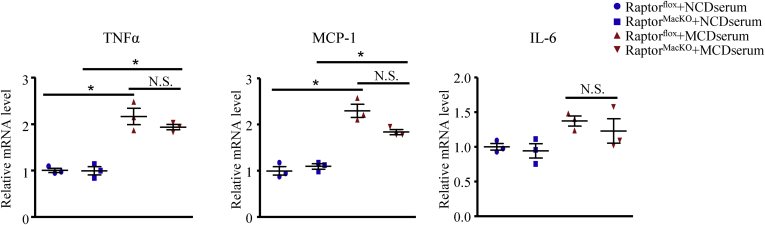
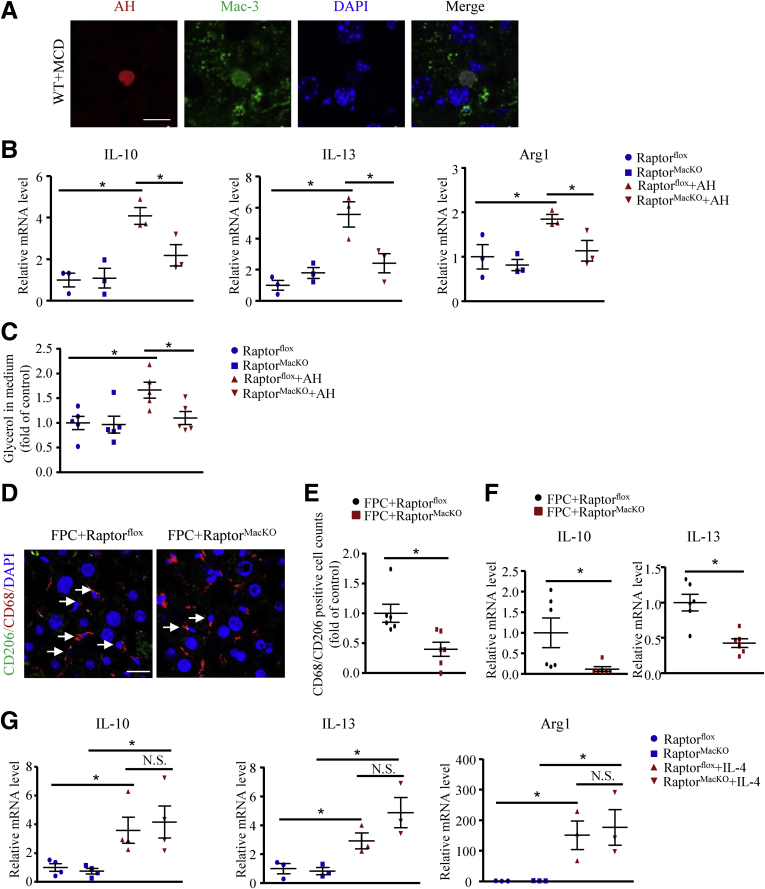
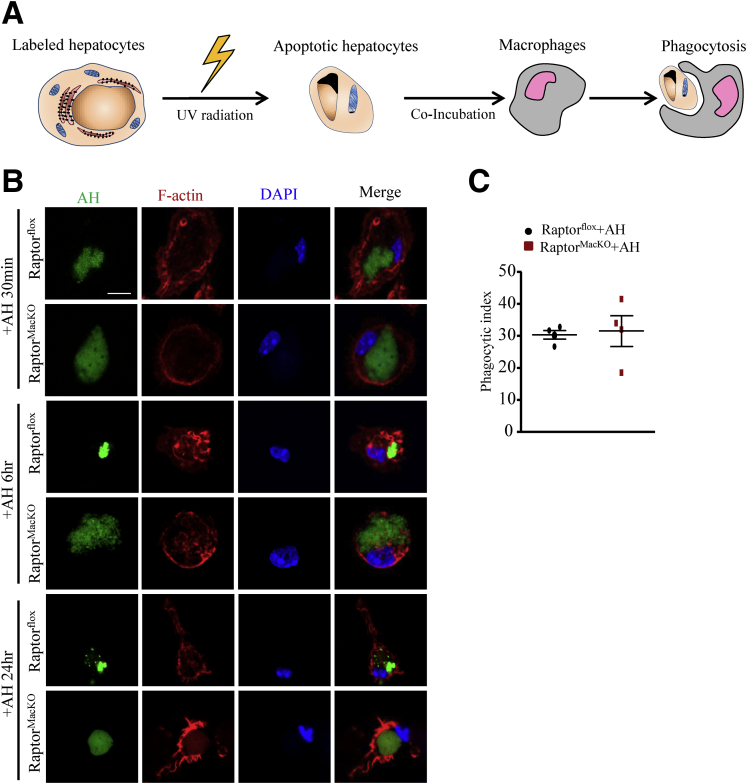
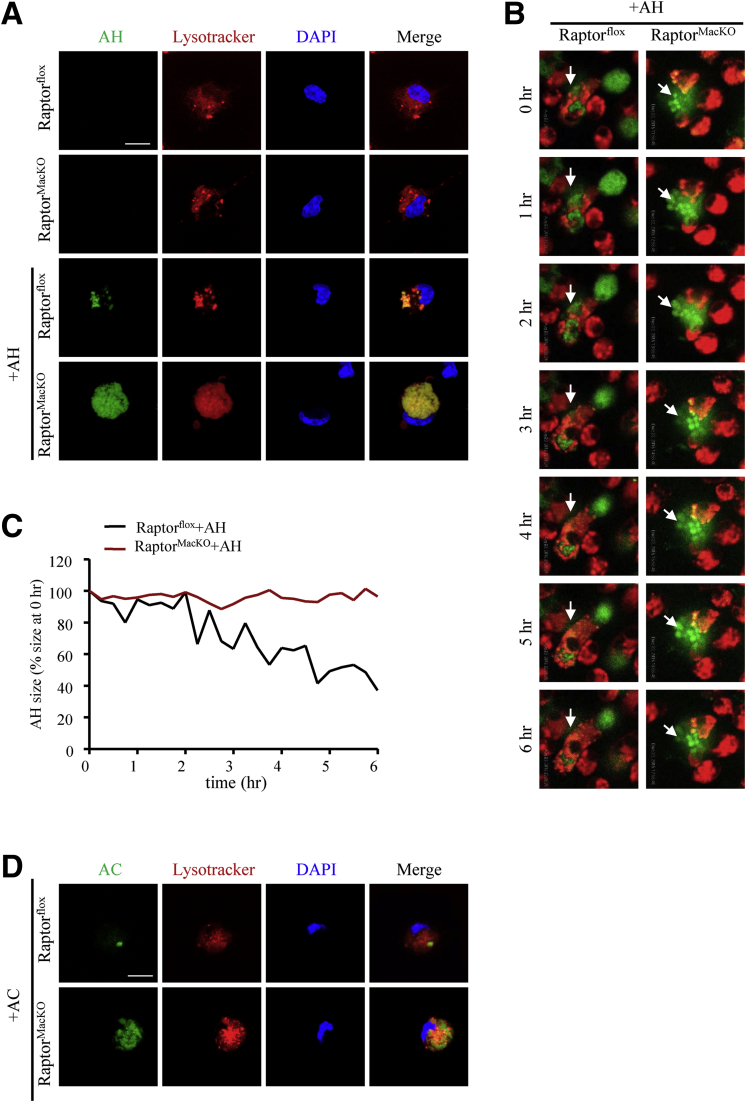
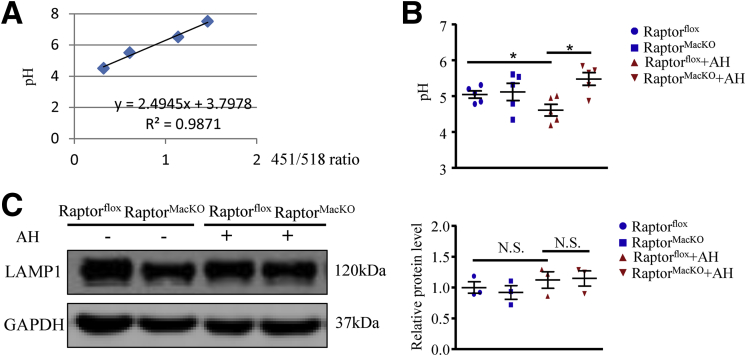
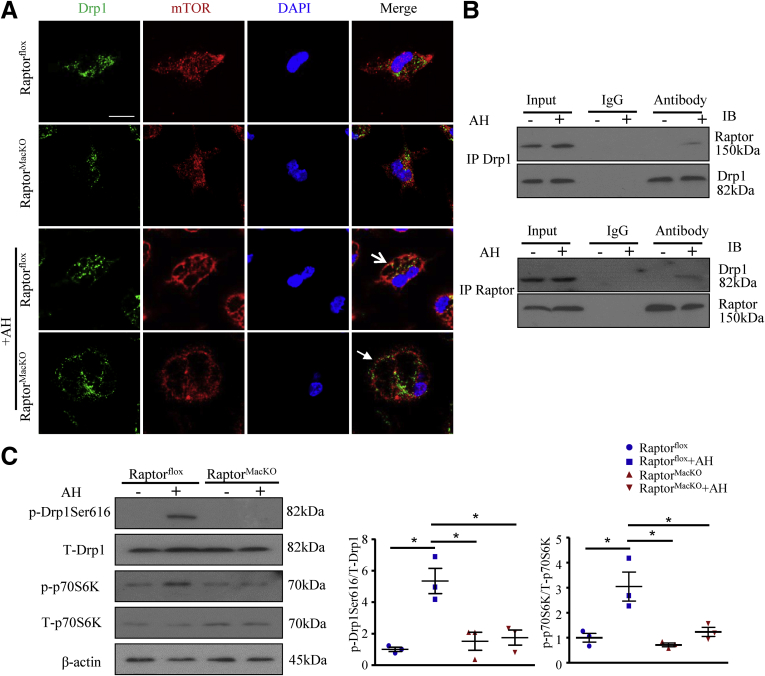
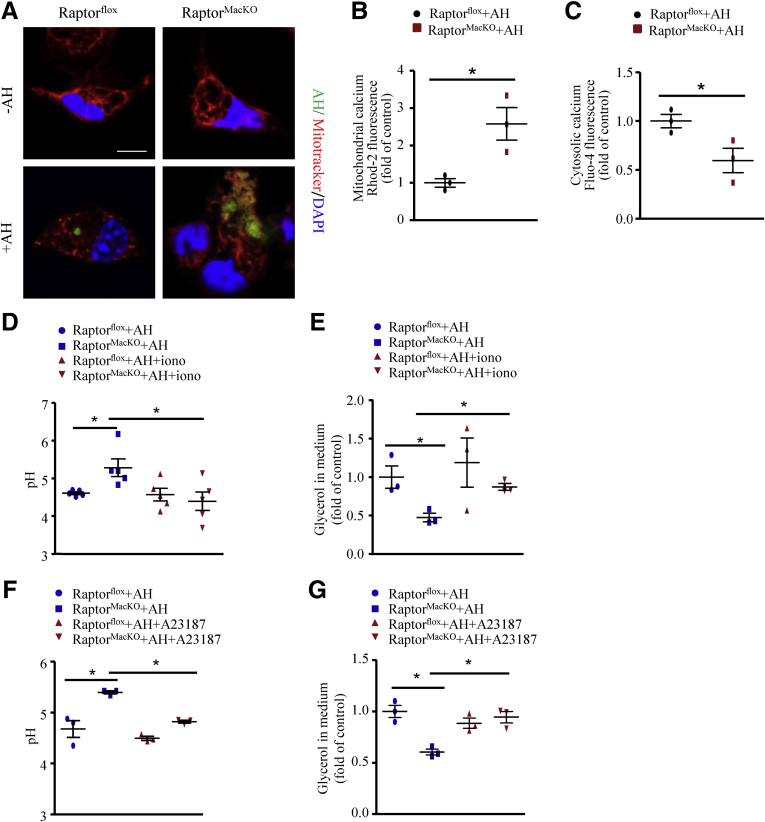
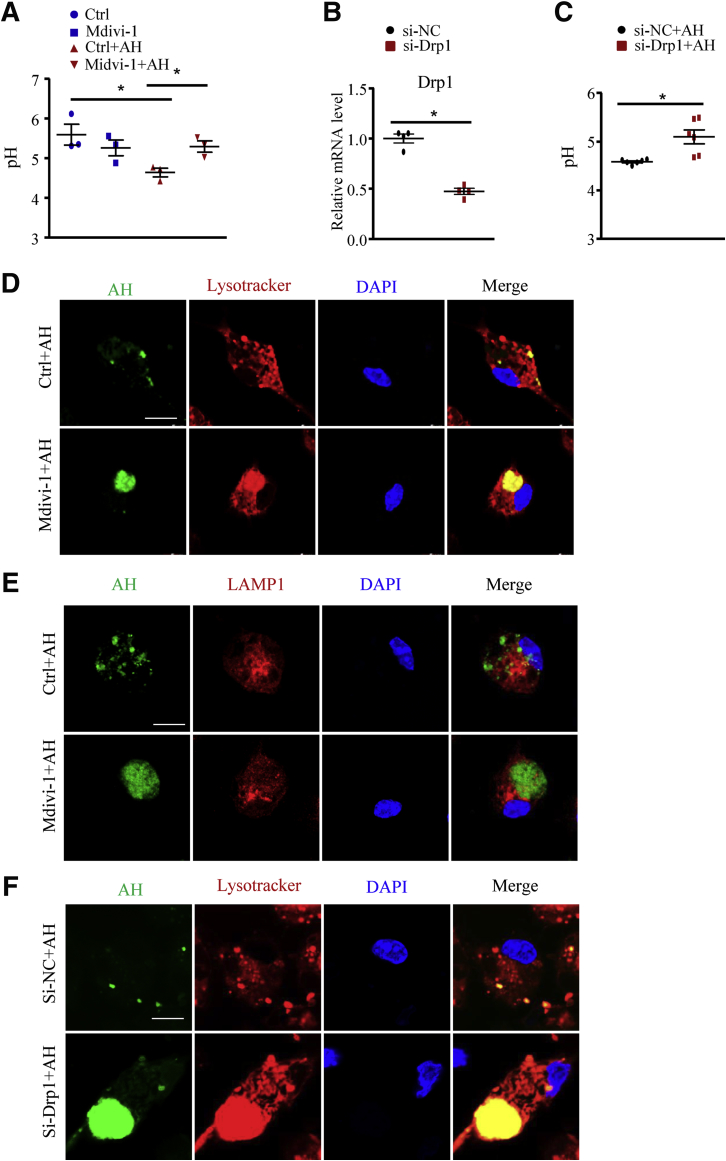
We report that in human beings bearing NASH, macrophage mTORC1 activity was lower in livers experiencing severe vs mild NASH liver. Moreover, macrophage mTORC1 disruption exacerbated the inflammatory response in 2 diet-induced NASH mouse models. Mechanistically, in response to apoptotic hepatocytes (AHs), macrophage polarization toward a M2 anti-inflammatory phenotype was inhibited in Raptor-deficient macrophages. During the digestion of AHs, macrophage mTORC1 was activated and coupled with dynamin-related protein 1 to facilitate the latter's phosphorylation, leading to mitochondrial fission-mediated calcium release. Ionomycin or A23187, calcium ionophores, prevented Raptor deficiency-mediated failure of lysosome acidification and subsequent lipolysis. Blocking dynamin-related protein 1-dependent mitochondria fission impaired lysosome function, resulting in reduced production of anti-inflammatory factors such as interleukins 10 and 13.
Persistent mTORC1 deficiency in macrophages contributes to the progression of NASH by causing lysosome dysfunction and subsequently attenuating anti-inflammatory M2-like response in macrophages during clearance of AHs.
非酒精性脂肪性肝炎(NASH)是一种日益流行的非酒精性脂肪性肝病,其特征为炎症细胞浸润和肝细胞损伤。雷帕霉素哺乳动物靶标(mTOR)复合物 1(mTORC1)在癌症的背景下,包括肝细胞癌,已经被广泛研究。然而,mTORC1 在 NASH 中的作用在很大程度上仍然未知。
比较了人类轻度和重度 NASH 肝脏中巨噬细胞的 mTORC1 活性。用高脂肪、棕榈酸和胆固醇饮食喂养具有巨噬细胞特异性 mTOR 调节相关蛋白(Raptor)亚基缺失的小鼠和同窝对照 24 周或蛋氨酸和胆碱缺乏饮食 4 周,以发展 NASH。
我们报告称,在患有 NASH 的人类中,与轻度 NASH 肝脏相比,经历重度 NASH 肝脏的巨噬细胞 mTORC1 活性较低。此外,在 2 种饮食诱导的 NASH 小鼠模型中,巨噬细胞 mTORC1 破坏加剧了炎症反应。从机制上讲,在响应凋亡的肝细胞(AHs)时,Raptor 缺陷型巨噬细胞中向抗炎的 M2 表型极化受到抑制。在 AHs 的消化过程中,巨噬细胞 mTORC1 被激活并与动力相关蛋白 1(dynamin-related protein 1,DRP1)偶联,以促进后者的磷酸化,从而导致线粒体分裂介导的钙释放。离子霉素或 A23187,钙载体,防止了 Raptor 缺陷介导的溶酶体酸化失败和随后的脂解。阻断依赖 dynamin-related protein 1 的线粒体分裂会损害溶酶体功能,导致抗炎因子如白细胞介素 10 和 13 的产生减少。
巨噬细胞中持续的 mTORC1 缺乏导致 NASH 的进展,原因是溶酶体功能障碍,随后在清除 AHs 期间减弱巨噬细胞中的抗炎 M2 样反应。