Department of Informatics, University of Oslo, Oslo, Norway.
K. G. Jebsen Coeliac Disease Research Centre, Oslo, Norway.
BMC Bioinformatics. 2018 Dec 14;19(1):481. doi: 10.1186/s12859-018-2438-1.
The current versions of reference genome assemblies still contain gaps represented by stretches of Ns. Since high throughput sequencing reads cannot be mapped to those gap regions, the regions are depleted of experimental data. Moreover, several technology platforms assay a targeted portion of the genomic sequence, meaning that regions from the unassayed portion of the genomic sequence cannot be detected in those experiments. We here refer to all such regions as inaccessible regions, and hypothesize that ignoring these regions in the null model may increase false findings in statistical testing of colocalization of genomic features.
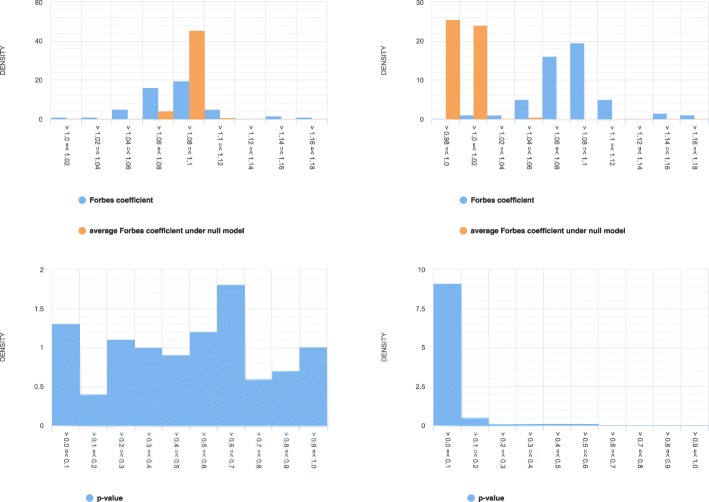
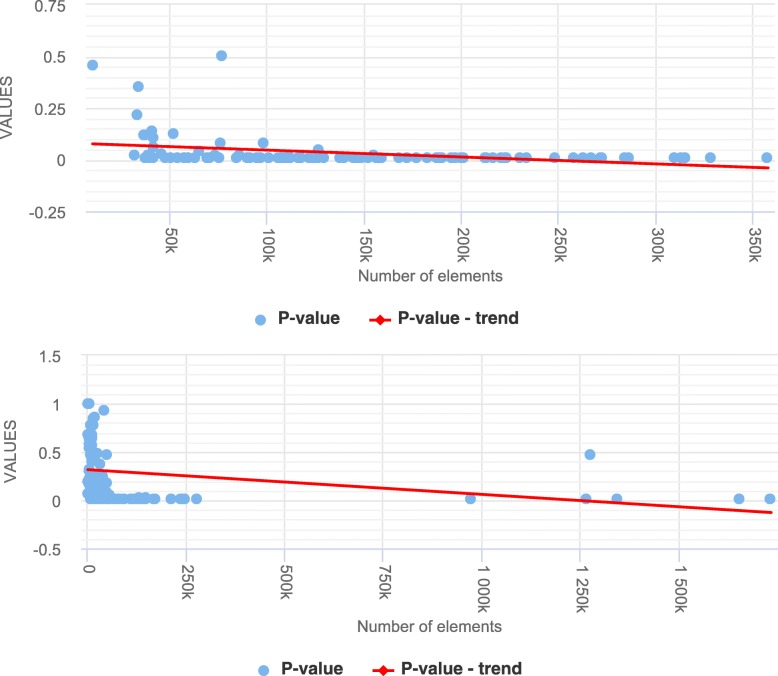
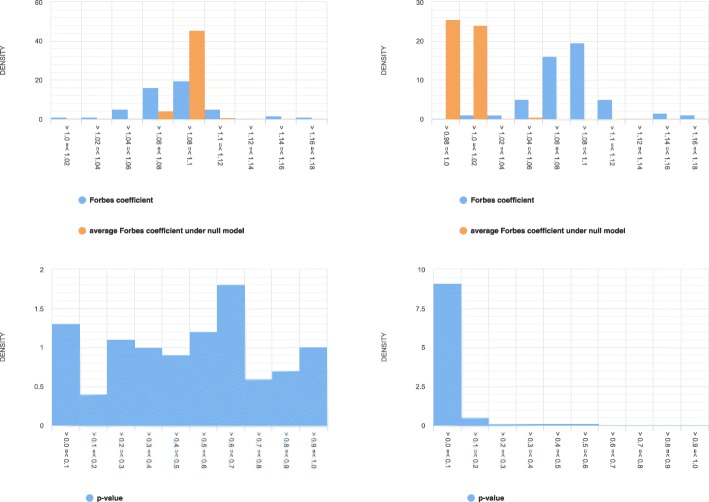
Our explorative analyses confirm that the genomic regions in public genomic tracks intersect very little with assembly gaps of human reference genomes (hg19 and hg38). The little intersection was observed only at the beginning and end portions of the gap regions. Further, we simulated a set of synthetic tracks by matching the properties of real genomic tracks in a way that nullified any true association between them. This allowed us to test our hypothesis that not avoiding inaccessible regions (as represented by assembly gaps) in the null model would result in spurious inflation of statistical significance. We contrasted the distributions of test statistics and p-values of Monte Carlo-based permutation tests that either avoided or did not avoid assembly gaps in the null model when testing colocalization between a pair of tracks. We observed that the statistical tests that did not account for assembly gaps in the null model resulted in a distribution of the test statistic that is shifted to the right and a distribution of p-values that is shifted to the left (indicating inflated significance). We observed a similar level of inflated significance in hg19 and hg38, despite assembly gaps covering a smaller proportion of the latter reference genome.
We provide empirical evidence demonstrating that inaccessible regions, even when covering only a few percentages of the genome, can lead to a substantial amount of false findings if not accounted for in statistical colocalization analysis.
当前版本的参考基因组组装仍然包含由 N 组成的大片段缺口。由于高通量测序reads 无法映射到这些缺口区域,这些区域缺乏实验数据。此外,有几种技术平台检测基因组序列的靶向部分,这意味着无法在这些实验中检测到基因组序列未检测部分的区域。我们将所有这些区域都称为不可及区域,并假设在统计测试中忽略这些区域的无效模型可能会增加基因组特征共定位的假阳性发现。
我们的探索性分析证实,公共基因组轨迹中的基因组区域与人类参考基因组(hg19 和 hg38)的组装缺口相交甚少。这种少量的交集仅在缺口区域的开始和结束部分观察到。此外,我们通过匹配真实基因组轨迹的属性来模拟一组合成轨迹,从而使它们之间的任何真实关联无效。这使我们能够测试我们的假设,即在无效模型中不避免不可及区域(如组装缺口所代表的)会导致统计显著性的虚假膨胀。我们对比了在测试一对轨迹之间的共定位时,无效模型中是否避免组装缺口的基于蒙特卡罗置换检验的检验统计量和 p 值的分布。我们观察到,在无效模型中不考虑组装缺口的统计检验导致检验统计量的分布向右移,p 值的分布向左移(表明显著性膨胀)。我们在 hg19 和 hg38 中观察到了类似水平的显著性膨胀,尽管后者参考基因组中的组装缺口仅覆盖了基因组的一小部分。
我们提供了经验证据,证明不可及区域,即使仅覆盖基因组的几个百分比,如果在统计共定位分析中不考虑这些区域,也可能导致大量的假阳性发现。