Peng Weijun, Yi Pengji, Yang Jingjing, Xu Panpan, Wang Yang, Zhang Zheyu, Huang Siqi, Wang Zhe, Zhang Chunhu
Department of Integrated Traditional Chinese & Western Medicine, The Second Xiangya Hospital, Central South University, Changsha, Hunan 410011, China.
Department of Integrated Traditional Chinese & Western Medicine, Xiangya Hospital, Central South University, Changsha, Hunan 410008, China.
Aging (Albany NY). 2018 Dec 18;10(12):4054-4065. doi: 10.18632/aging.101693.
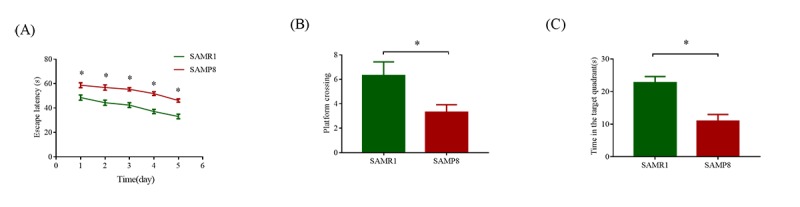
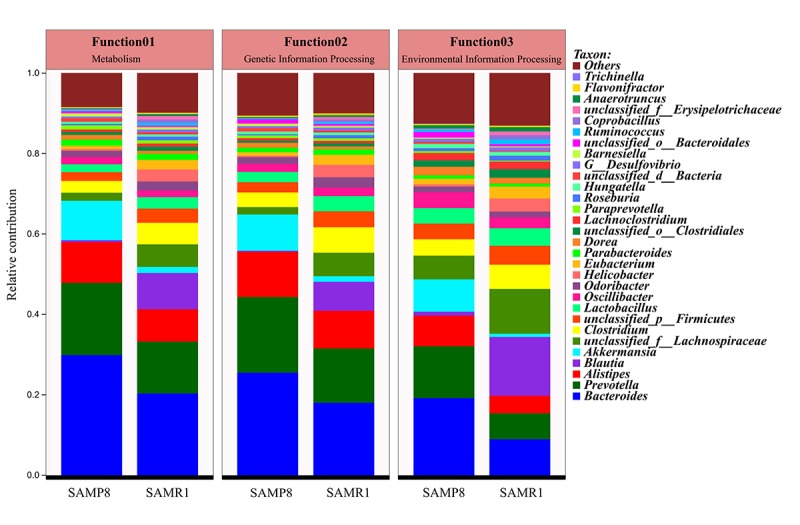
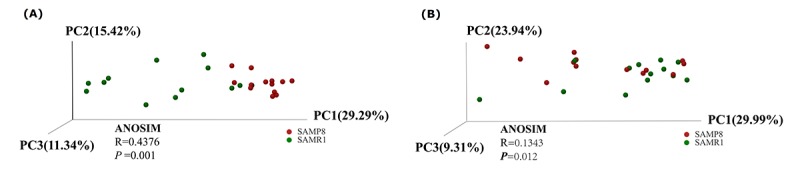
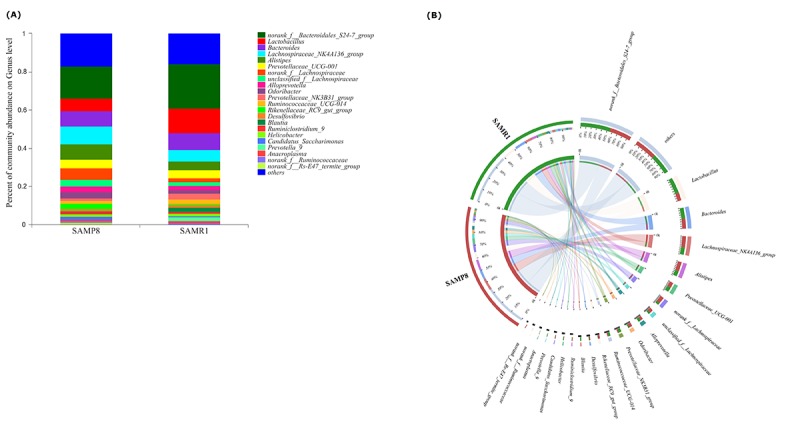
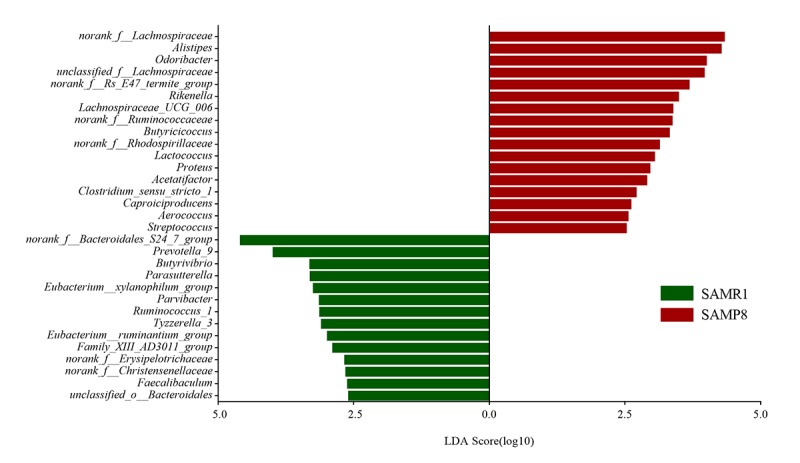
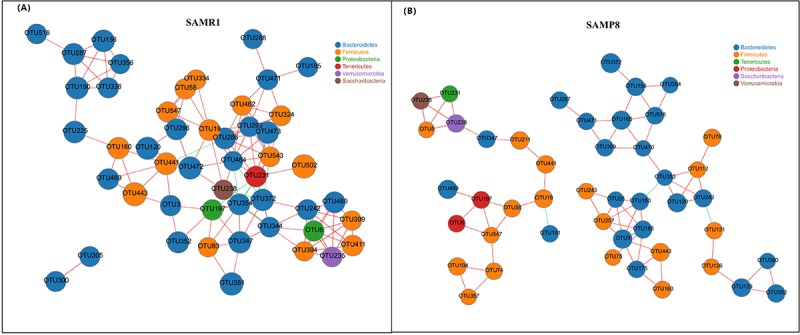
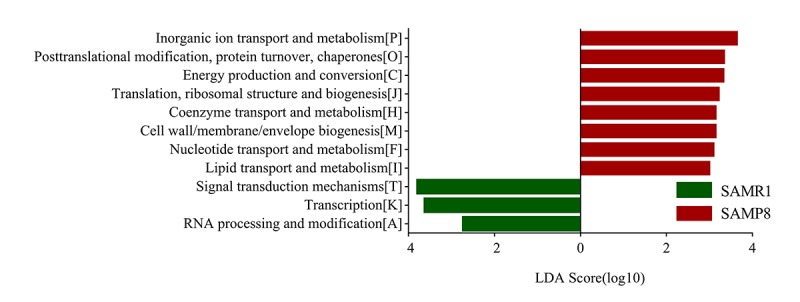
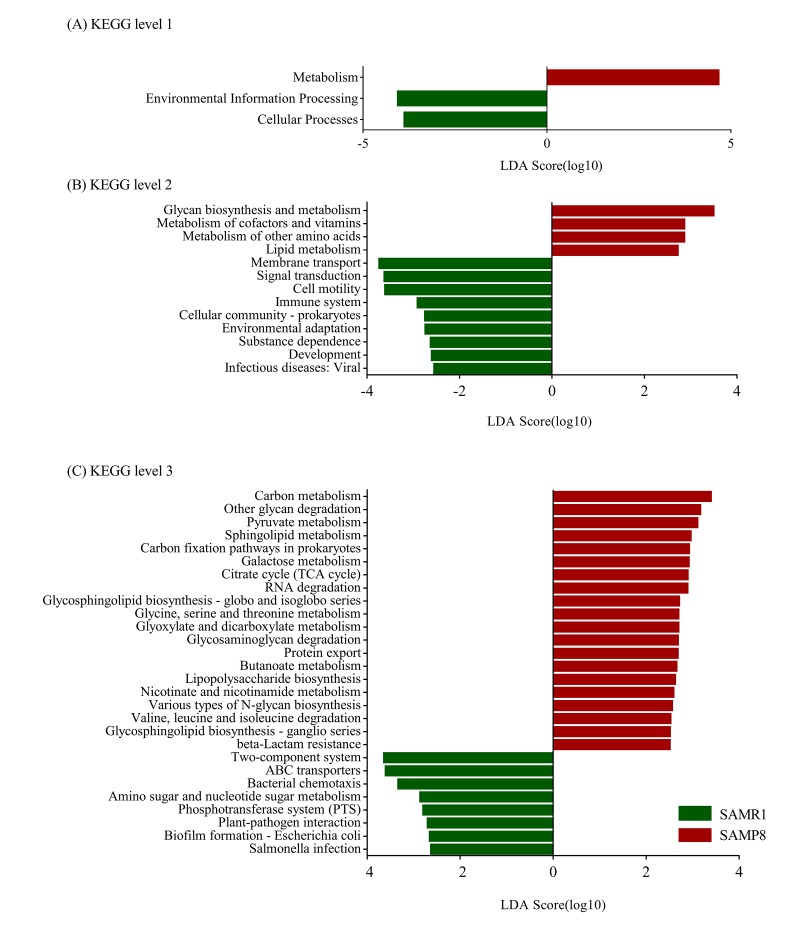
Although an intriguing potential association of the gut microbiome with Alzheimer's disease (AD) has attracted recent interest, few studies have directly assessed this relationship or underlying mechanism. Here, we compared the gut microbiota composition and functional differentiation of senescence-accelerated mouse prone 8 (SAMP8) mice with control senescence-accelerated mouse resistant 1 (SAMR1) mice using 16S rRNA gene and metagenomic sequencing analysis, respectively. Specifically, 16S sequencing results showed that the SAMP8 mice displayed a characteristic composition of the gut microbiome that clearly differed from that of the SAMR1 mice. Moreover, network analysis revealed that the gut microbiota of SAMP8 mice had decreased correlation density and clustering of operational taxonomic units. Metagenomic results revealed that the predominant Cluster of Orthologous Groups functional category related to these changes was the metabolism cluster in SAMP8 mice. The Kyoto Encyclopedia of Genes and Genomes (KEGG) annotation further demonstrated enrichment of the relative abundance of some dominant metabolism-related KEGG pathways in the SAMP8 mice, consistent with the suggested pathogenic mechanisms of AD. In conclusion, this study suggests that perturbations of the gut microbiota composition and the functional metagenome may be associated with AD. Further studies are warranted to elucidate the potential new mechanism contributing to AD progression.
尽管肠道微生物群与阿尔茨海默病(AD)之间存在潜在关联这一有趣现象最近引起了人们的关注,但很少有研究直接评估这种关系或潜在机制。在此,我们分别使用16S rRNA基因和宏基因组测序分析,比较了快速老化小鼠易感8型(SAMP8)小鼠与对照快速老化小鼠抗性1型(SAMR1)小鼠的肠道微生物群组成和功能分化。具体而言,16S测序结果表明,SAMP8小鼠呈现出与SAMR1小鼠明显不同的肠道微生物群特征组成。此外,网络分析显示,SAMP8小鼠的肠道微生物群中,操作分类单元的相关密度和聚类有所降低。宏基因组结果显示,与这些变化相关的主要直系同源基因簇功能类别是SAMP8小鼠中的代谢簇。京都基因与基因组百科全书(KEGG)注释进一步证明,SAMP8小鼠中一些主要代谢相关KEGG通路的相对丰度有所富集,这与AD的推测致病机制一致。总之,本研究表明肠道微生物群组成和功能宏基因组的扰动可能与AD有关。有必要进一步开展研究,以阐明导致AD进展的潜在新机制。