Loucks Alexandra D, O'Hara Thomas, Trayanova Natalia A
Institute for Computational Medicine and Department of Biomedical Engineering at Johns Hopkins University, Baltimore, MD, United States.
Front Physiol. 2018 Dec 4;9:1737. doi: 10.3389/fphys.2018.01737. eCollection 2018.
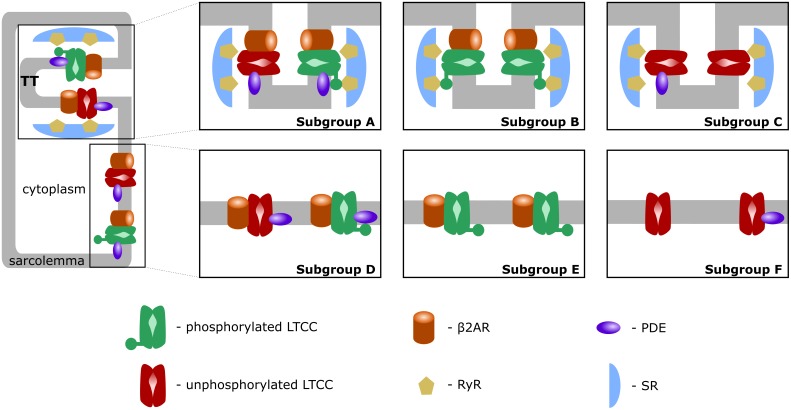
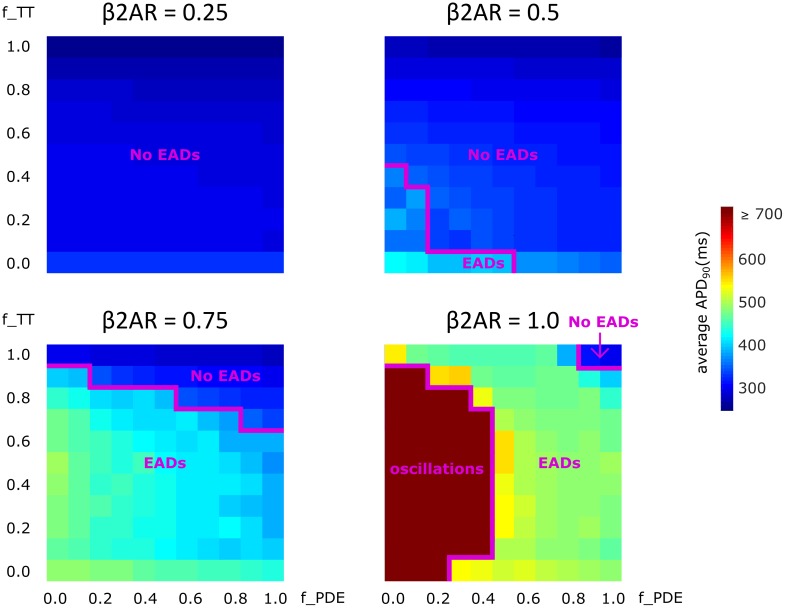
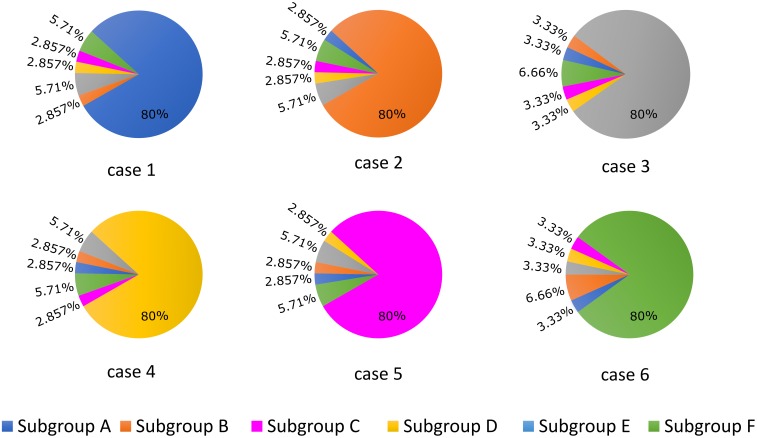
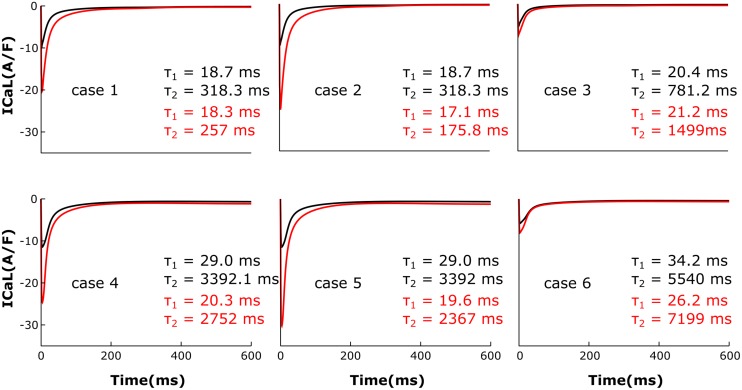
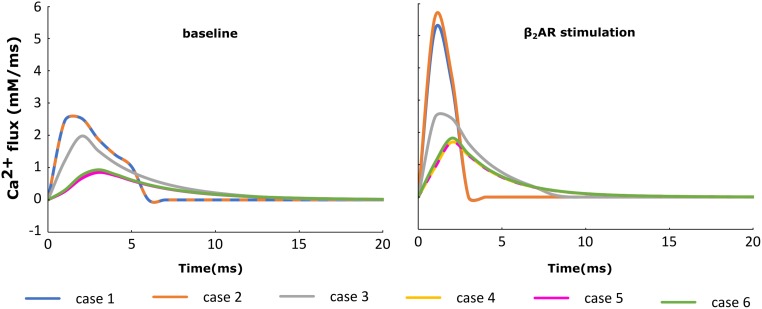
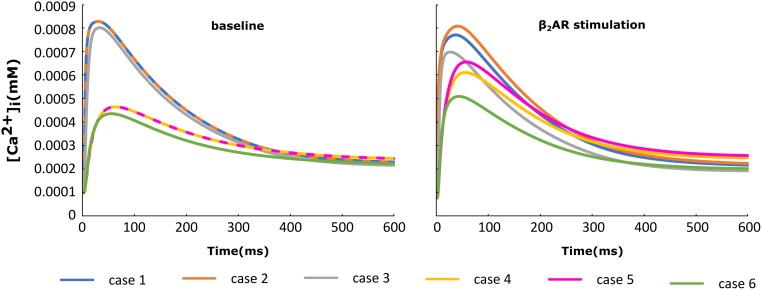
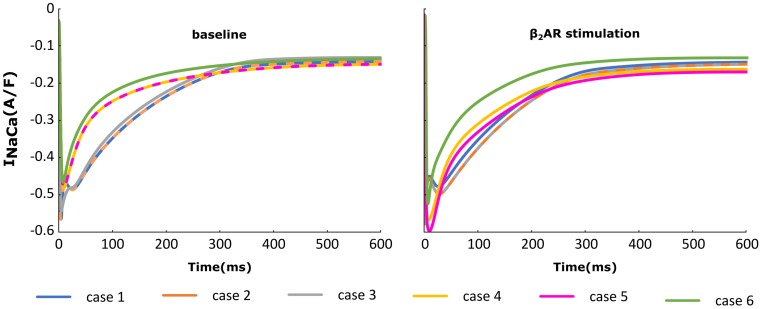
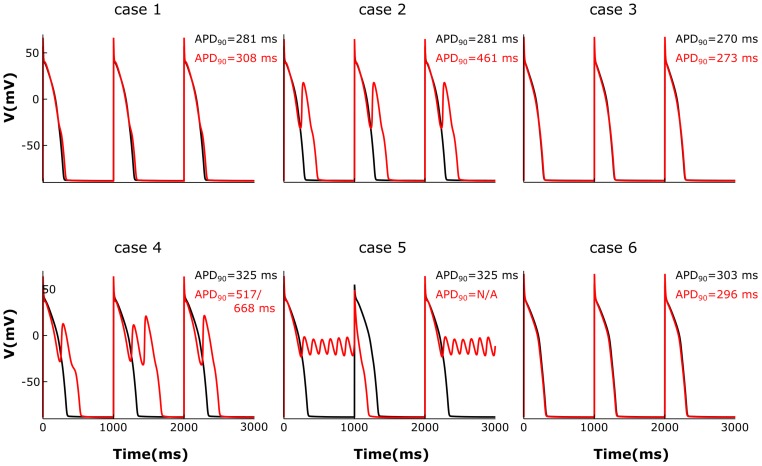
Heart failure (HF) is one of the most common causes of morbidity and mortality worldwide. Although many patients suffering from HF die from sudden cardiac death caused by arrhythmias, the mechanism linking HF remodeling to an increased arrhythmogenic propensity remains incomplete. HF is typically characterized by a progressive loss of transverse tubule (T-tubule) domains, which leads to an altered distribution of L-type calcium channels (LTCCs). Microdomain degradation also causes the disruption of the β adrenergic receptor (βAR) and phosphodiesterase (PDE) signaling localization, normally confined to the dyadic space. The goal of this study was to analyze how these subcellular changes affect the function of LTCCs and lead to the emergence of ventricular cell-level triggers of arrhythmias. To accomplish this, we developed a novel computational model of a human ventricular HF myocyte in which LTCCs were divided into six different populations, based on their location and signaling environment they experience. To do so, we included T-tubular microdomain remodeling which led to a subset of LTCCs to be redistributed from the T-tubular to the surface membrane and allowed for different levels of phosphorylation of LTCCs by PKA, based on the presence of βARs and PDEs. The model was used to study the behavior of the LTCC current (I) under basal and sympathetic stimulation and its effect on cellular action potential. Our results showed that channels redistributed from the T-tubular membrane to the bulk of the sarcolemma displayed an altered function in their new, non-native signaling domain. Incomplete calcium dependent inactivation, which resulted in a longer-lasting and larger-in-magnitude LTCC current, was observed when we decoupled LTCCs from ryanodine receptors and removed them from the dyadic space. The magnitude of the LTCC current, especially in the surface sarcolemma, was also increased via phosphorylation by the redistributed βARs and PDEs. These changes in LTCC current led to the development of early afterdepolarizations. Thus, our study shows that altered LTCC function is a potential cause for the emergence of cell-level triggers of arrhythmia, and that βARs and PDEs present useful therapeutic targets for treatment of HF and prevention of sudden cardiac death.
心力衰竭(HF)是全球发病和死亡的最常见原因之一。尽管许多HF患者死于心律失常导致的心脏性猝死,但将HF重塑与心律失常倾向增加联系起来的机制仍不完整。HF的典型特征是横管(T管)结构域逐渐丧失,这导致L型钙通道(LTCCs)分布改变。微结构域降解还会导致β肾上腺素能受体(βAR)和磷酸二酯酶(PDE)信号定位的破坏,这些信号通常局限于二联体空间。本研究的目的是分析这些亚细胞变化如何影响LTCCs的功能,并导致心室细胞水平心律失常触发因素的出现。为了实现这一目标,我们开发了一种新型的人类心室HF心肌细胞计算模型,其中LTCCs根据其位置和所经历的信号环境被分为六个不同的群体。为此,我们纳入了T管微结构域重塑,这导致一部分LTCCs从T管重新分布到表面膜,并根据βARs和PDEs的存在情况允许LTCCs被蛋白激酶A(PKA)进行不同程度的磷酸化。该模型用于研究基础和交感神经刺激下LTCC电流(I)的行为及其对细胞动作电位的影响。我们的结果表明,从T管膜重新分布到肌膜大部分区域的通道在其新的、非天然的信号结构域中表现出功能改变。当我们将LTCCs与兰尼碱受体解耦并将它们从二联体空间中移除时,观察到不完全的钙依赖性失活,这导致LTCC电流持续时间更长、幅度更大。重新分布的βARs和PDEs通过磷酸化也增加了LTCC电流的幅度,尤其是在表面肌膜中。LTCC电流的这些变化导致了早期后去极化的发生。因此,我们的研究表明,LTCC功能改变是心律失常细胞水平触发因素出现的潜在原因,并且βARs和PDEs是治疗HF和预防心脏性猝死的有用治疗靶点。