Morgan Andrew J, Ayyer Kartik, Barty Anton, Chen Joe P J, Ekeberg Tomas, Oberthuer Dominik, White Thomas A, Yefanov Oleksandr, Chapman Henry N
Center for Free-Electron Laser Science, Deutsches Elektronen-Synchrotron DESY, Notkestrasse 85, 22607 Hamburg, Germany.
Department of Physics, Arizona State University, Tempe, AZ, 85287, USA.
Acta Crystallogr A Found Adv. 2019 Jan 1;75(Pt 1):25-40. doi: 10.1107/S2053273318015395.
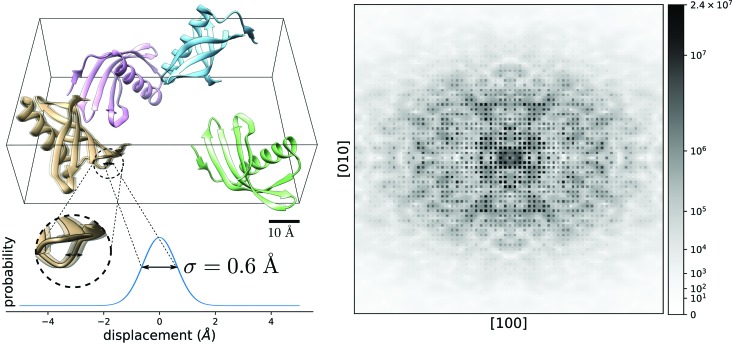
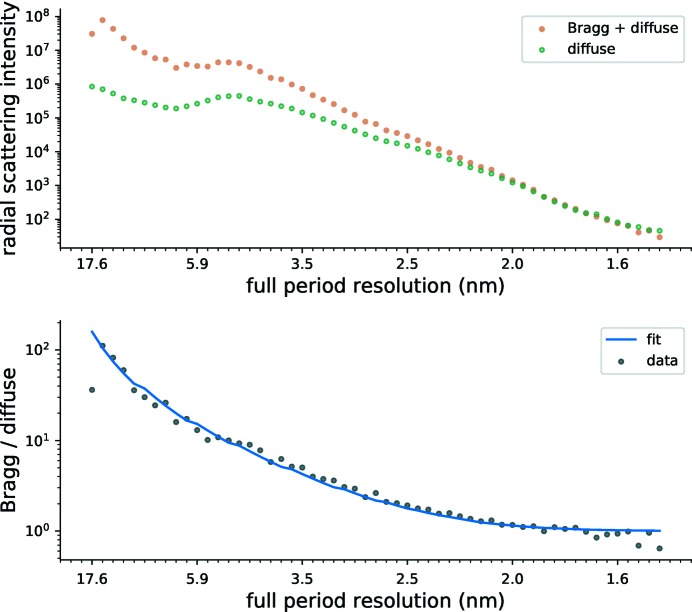
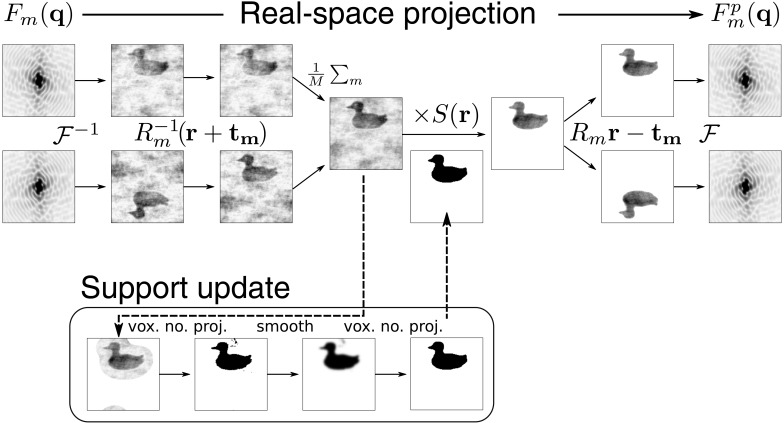
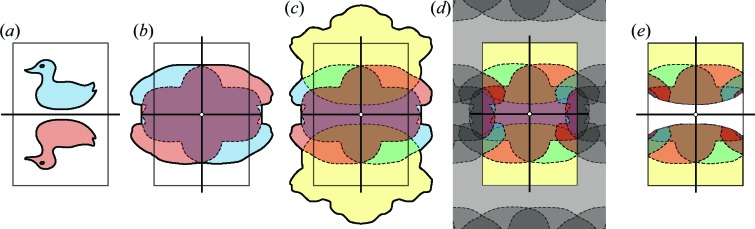
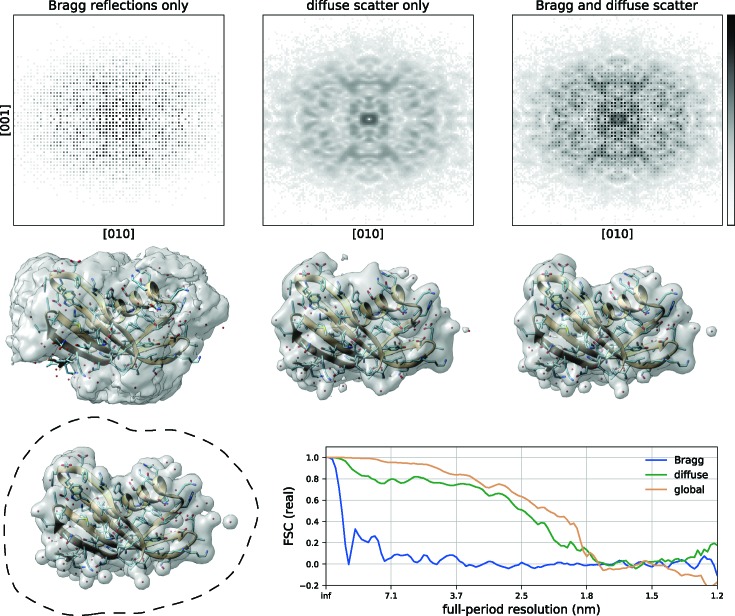
To date X-ray protein crystallography is the most successful technique available for the determination of high-resolution 3D structures of biological molecules and their complexes. In X-ray protein crystallography the structure of a protein is refined against the set of observed Bragg reflections from a protein crystal. The resolution of the refined protein structure is limited by the highest angle at which Bragg reflections can be observed. In addition, the Bragg reflections alone are typically insufficient (by a factor of two) to determine the structure ab initio, and so prior information is required. Crystals formed from an imperfect packing of the protein molecules may also exhibit continuous diffraction between and beyond these Bragg reflections. When this is due to random displacements of the molecules from each crystal lattice site, the continuous diffraction provides the necessary information to determine the protein structure without prior knowledge, to a resolution that is not limited by the angular extent of the observed Bragg reflections but instead by that of the diffraction as a whole. This article presents an iterative projection algorithm that simultaneously uses the continuous diffraction as well as the Bragg reflections for the determination of protein structures. The viability of this method is demonstrated on simulated crystal diffraction.
迄今为止,X射线蛋白质晶体学是用于确定生物分子及其复合物高分辨率三维结构的最成功技术。在X射线蛋白质晶体学中,蛋白质的结构是根据从蛋白质晶体观察到的一组布拉格反射进行精修的。精修后的蛋白质结构分辨率受可观察到布拉格反射的最高角度限制。此外,仅布拉格反射通常不足以(相差两倍)从头确定结构,因此需要先验信息。由蛋白质分子不完全堆积形成的晶体在这些布拉格反射之间及之外也可能表现出连续衍射。当这是由于分子从每个晶格位点的随机位移导致时,连续衍射提供了无需先验知识即可确定蛋白质结构的必要信息,其分辨率不受观察到的布拉格反射角度范围的限制,而是受整个衍射的角度范围限制。本文提出了一种迭代投影算法,该算法同时使用连续衍射和布拉格反射来确定蛋白质结构。该方法的可行性在模拟晶体衍射上得到了证明。