Filosto Massimiliano, Piccinelli Stefano Cotti, Palmieri Ilaria, Necchini Nicola, Valente Marialuisa, Zanella Isabella, Biasiotto Giorgio, Lorenzo Diego Di, Cereda Cristina, Padovani Alessandro
Center for Neuromuscular Diseases, Unit of Neurology, ASST Spedali Civili and University of Brescia, 25100 Brescia, Italy.
Genomic and Post-Genomic Center, IRCCS Mondino Fundation, 27100 Pavia, Italy.
J Clin Med. 2018 Dec 22;8(1):17. doi: 10.3390/jcm8010017.
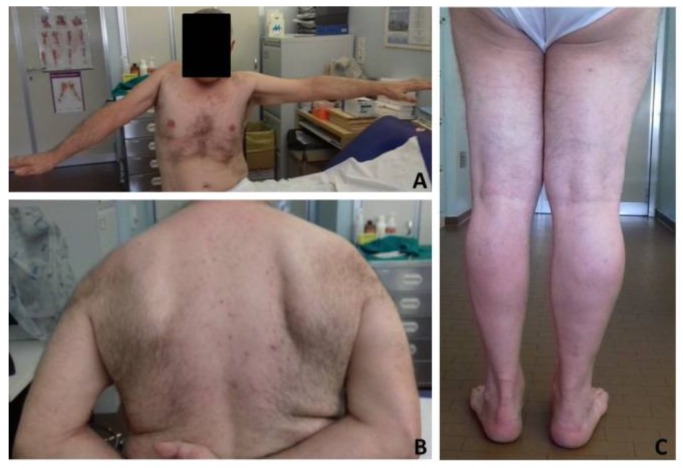
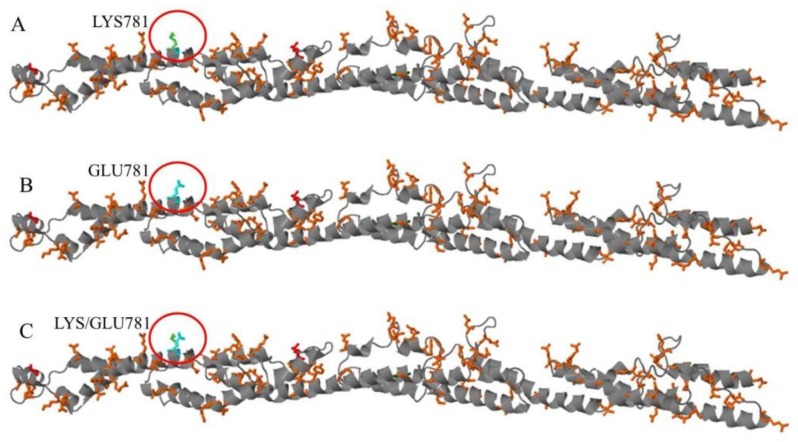
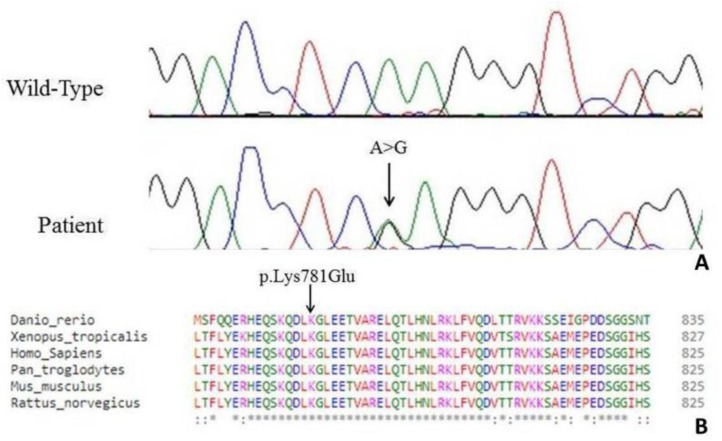
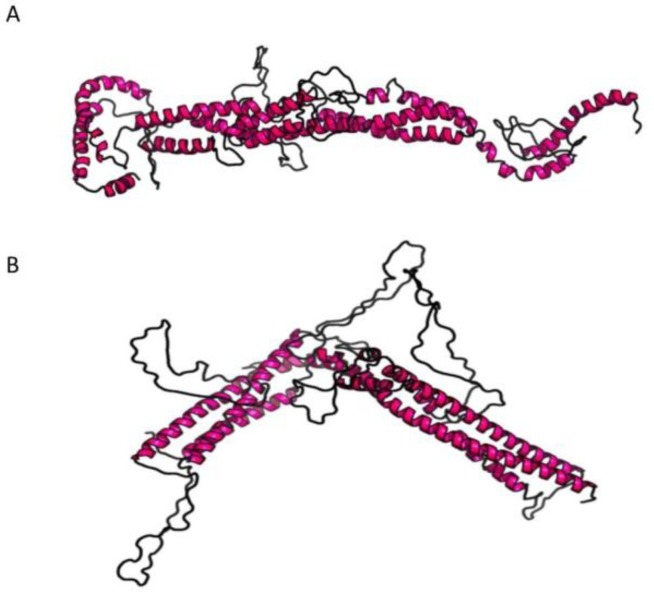
encodes the heavy chain A of kinesin; A motor protein involved in motility functions within neuron. Mutations in the N-terminal motor domain are known to cause SPG10; An autosomal dominant hereditary spastic paraplegia (HSP), as well as rare Charcot-Marie-Tooth disease 2 (CMT2) cases. Recently C-terminal cargo-binding tail domain mutations have been associated with an amyotrophic lateral sclerosis (ALS) phenotype. Here we describe a subject presenting with an atypical slowly progressive motor syndrome evolving over a period of 4 years; Characterized by walking difficulties; Muscle hypotrophy mainly involving upper limbs and pyramidal signs confined to the lower limbs. Electromyography demonstrated chronic neurogenic damage and active denervation while electroneurography showed slowly worsening axonal damage. We identified the novel heterozygote variant c.2341A>G in the exon 21 of the gene resulting in the amino acid change p.Lys781Glu. The residue Lys781 is located within the terminal region of the stalk domain and is highly evolutionary conserved. Our findings confirm that mutations in cause ALS-like phenotypes. However, the stalk domain mutation described here appears to result in an "intermediate" slowly progressive phenotype having aspects resembling ALS as well as HSP and axonal neuropathy. We suggest that gene should be considered as a candidate gene in all atypical progressive motor syndromes.
编码驱动蛋白重链A;一种参与神经元运动功能的运动蛋白。已知N端运动结构域的突变会导致SPG10;一种常染色体显性遗传性痉挛性截瘫(HSP),以及罕见的遗传性运动感觉神经病2型(CMT2)病例。最近,C端货物结合尾结构域的突变与肌萎缩侧索硬化症(ALS)表型有关。在此,我们描述了一名患者,其表现为一种非典型的缓慢进展性运动综合征,病程长达4年;其特征为行走困难;肌肉萎缩主要累及上肢,锥体束征局限于下肢。肌电图显示慢性神经源性损伤和活动性失神经,而神经电图显示轴索性损伤逐渐加重。我们在该基因的第21外显子中鉴定出新型杂合变异c.2341A>G,导致氨基酸变化p.Lys781Glu。赖氨酸781残基位于柄结构域的末端区域,且具有高度进化保守性。我们的研究结果证实,该基因的突变会导致ALS样表型。然而,此处描述的柄结构域突变似乎导致了一种“中间型”缓慢进展性表型,具有类似于ALS以及HSP和轴索性神经病的特征。我们建议,在所有非典型进行性运动综合征中,该基因都应被视为候选基因。