Bris Céline, Goudenege David, Desquiret-Dumas Valérie, Charif Majida, Colin Estelle, Bonneau Dominique, Amati-Bonneau Patrizia, Lenaers Guy, Reynier Pascal, Procaccio Vincent
UMR CNRS 6015-INSERM U1083, MitoVasc Institute, Angers University, Angers, France.
Biochemistry and Genetics Department, Angers Hospital, Angers, France.
Front Genet. 2018 Dec 11;9:632. doi: 10.3389/fgene.2018.00632. eCollection 2018.
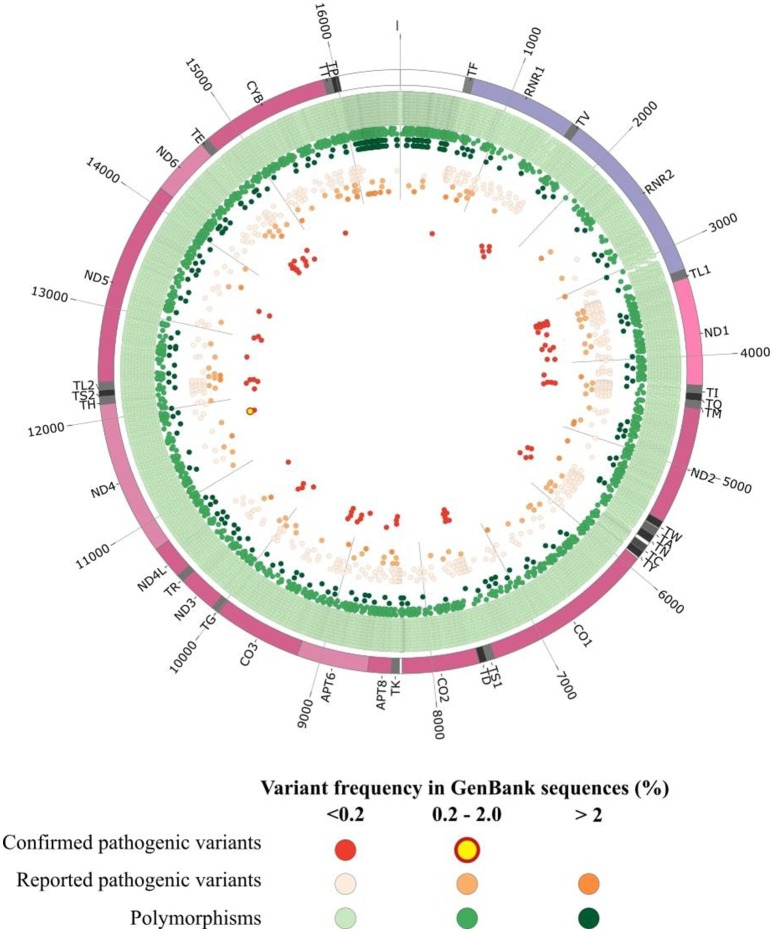
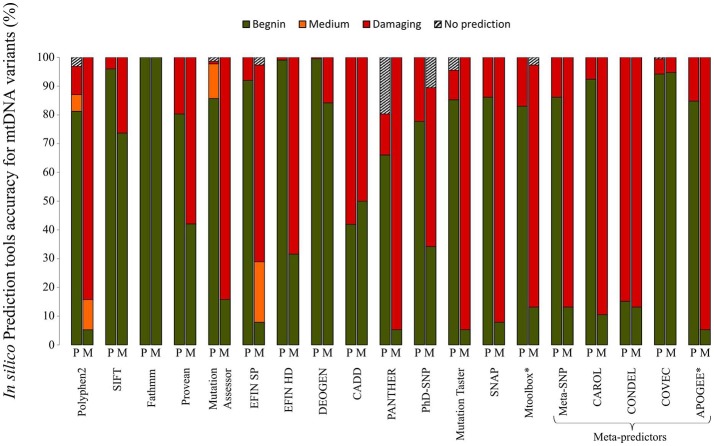
The development of next generation sequencing (NGS) has greatly enhanced the diagnosis of mitochondrial disorders, with a systematic analysis of the whole mitochondrial DNA (mtDNA) sequence and better detection sensitivity. However, the exponential growth of sequencing data renders complex the interpretation of the identified variants, thereby posing new challenges for the molecular diagnosis of mitochondrial diseases. Indeed, mtDNA sequencing by NGS requires specific bioinformatics tools and the adaptation of those developed for nuclear DNA, for the detection and quantification of mtDNA variants from sequence alignment to the calling steps, in order to manage the specific features of the mitochondrial genome including heteroplasmy, i.e., coexistence of mutant and wildtype mtDNA copies. The prioritization of mtDNA variants remains difficult, relying on a limited number of specific resources: population and clinical databases, and tools providing a prediction of the variant pathogenicity. An evaluation of the most prominent bioinformatics tools showed that their ability to predict the pathogenicity was highly variable indicating that special efforts should be directed at developing new bioinformatics tools dedicated to the mitochondrial genome. In addition, massive parallel sequencing raised several issues related to the interpretation of very low mtDNA mutational loads, discovery of variants of unknown significance, and mutations unrelated to patient phenotype or the co-occurrence of mtDNA variants. This review provides an overview of the current strategies and bioinformatics tools for accurate annotation, prioritization and reporting of mtDNA variations from NGS data, in order to carry out accurate genetic counseling in individuals with primary mitochondrial diseases.
下一代测序(NGS)技术的发展极大地促进了线粒体疾病的诊断,它能够对整个线粒体DNA(mtDNA)序列进行系统分析,检测灵敏度也更高。然而,测序数据的指数级增长使得对已识别变异的解读变得复杂,从而给线粒体疾病的分子诊断带来了新的挑战。实际上,通过NGS进行mtDNA测序需要特定的生物信息学工具,以及对那些为核DNA开发的工具进行调整,以便从序列比对到变异位点识别步骤来检测和定量mtDNA变异,从而应对线粒体基因组的特殊特征,包括异质性,即突变型和野生型mtDNA拷贝的共存。mtDNA变异的优先级排序仍然很困难,这依赖于有限的特定资源:人群和临床数据库,以及能够预测变异致病性的工具。对最突出的生物信息学工具的评估表明,它们预测致病性的能力差异很大,这表明应特别致力于开发专门针对线粒体基因组的新生物信息学工具。此外,大规模平行测序引发了几个与极低mtDNA突变负荷的解读、未知意义变异的发现、与患者表型无关的突变或mtDNA变异的共现相关的问题。本综述概述了当前用于准确注释、优先排序和报告来自NGS数据的mtDNA变异的策略和生物信息学工具,以便对原发性线粒体疾病患者进行准确的遗传咨询。