Section of Genetic Medicine, The University of Chicago, Chicago, Illinois, United States of America.
Department of Biology, Loyola University Chicago, Chicago, Illinois, United States of America.
PLoS Genet. 2019 Jan 22;15(1):e1007889. doi: 10.1371/journal.pgen.1007889. eCollection 2019 Jan.
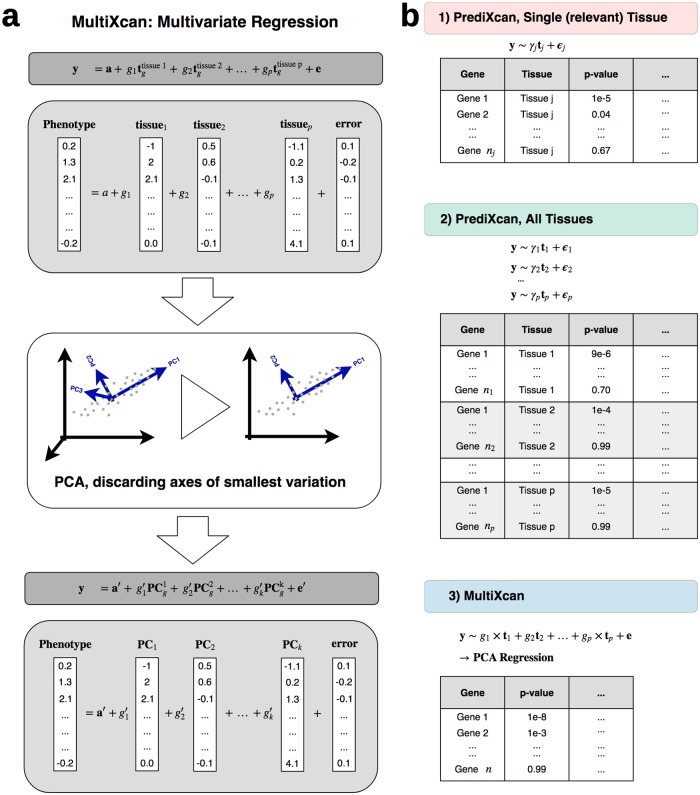
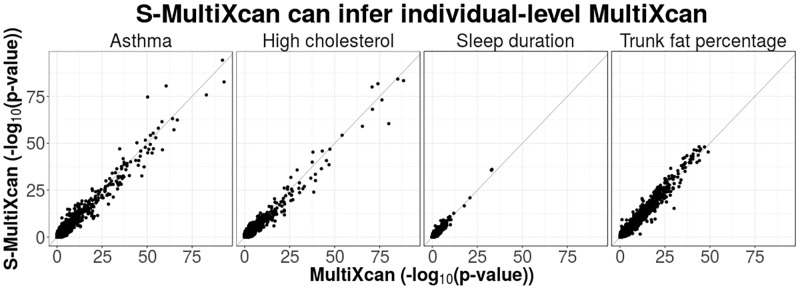
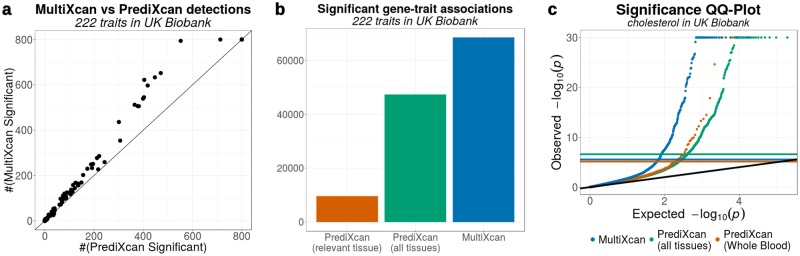
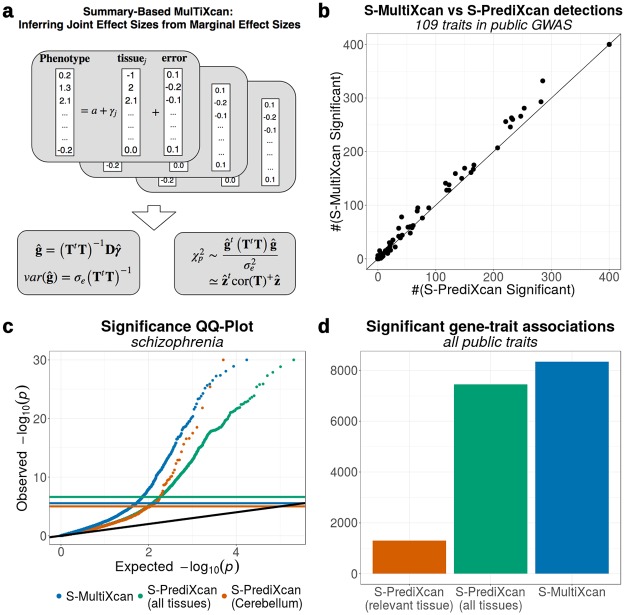
Integration of genome-wide association studies (GWAS) and expression quantitative trait loci (eQTL) studies is needed to improve our understanding of the biological mechanisms underlying GWAS hits, and our ability to identify therapeutic targets. Gene-level association methods such as PrediXcan can prioritize candidate targets. However, limited eQTL sample sizes and absence of relevant developmental and disease context restrict our ability to detect associations. Here we propose an efficient statistical method (MultiXcan) that leverages the substantial sharing of eQTLs across tissues and contexts to improve our ability to identify potential target genes. MultiXcan integrates evidence across multiple panels using multivariate regression, which naturally takes into account the correlation structure. We apply our method to simulated and real traits from the UK Biobank and show that, in realistic settings, we can detect a larger set of significantly associated genes than using each panel separately. To improve applicability, we developed a summary result-based extension called S-MultiXcan, which we show yields highly concordant results with the individual level version when LD is well matched. Our multivariate model-based approach allowed us to use the individual level results as a gold standard to calibrate and develop a robust implementation of the summary-based extension. Results from our analysis as well as software and necessary resources to apply our method are publicly available.
需要整合全基因组关联研究(GWAS)和表达数量性状基因座(eQTL)研究,以提高我们对 GWAS 命中背后的生物学机制的理解,以及我们识别治疗靶点的能力。基因水平关联方法,如 PrediXcan,可以优先考虑候选靶点。然而,有限的 eQTL 样本量和缺乏相关的发育和疾病背景限制了我们检测关联的能力。在这里,我们提出了一种有效的统计方法(MultiXcan),该方法利用组织和背景之间大量共享的 eQTL 来提高我们识别潜在靶基因的能力。MultiXcan 使用多元回归整合多个面板的证据,自然考虑了相关性结构。我们将我们的方法应用于来自英国生物库的模拟和真实特征,并表明,在现实环境中,我们可以检测到更多与基因显著相关的基因,而不是单独使用每个面板。为了提高适用性,我们开发了一种基于汇总结果的扩展,称为 S-MultiXcan,当 LD 匹配良好时,我们表明该扩展与个体水平版本具有高度一致性。我们的基于多元模型的方法允许我们使用个体水平的结果作为金标准来校准和开发基于汇总的扩展的稳健实现。我们的分析结果以及应用我们方法所需的软件和必要资源都是公开的。