Section of Genetic Medicine, The University of Chicago, Chicago, IL, 60637, USA.
Department of Biology, Loyola University Chicago, Chicago, IL, 60660, USA.
Nat Commun. 2018 May 8;9(1):1825. doi: 10.1038/s41467-018-03621-1.
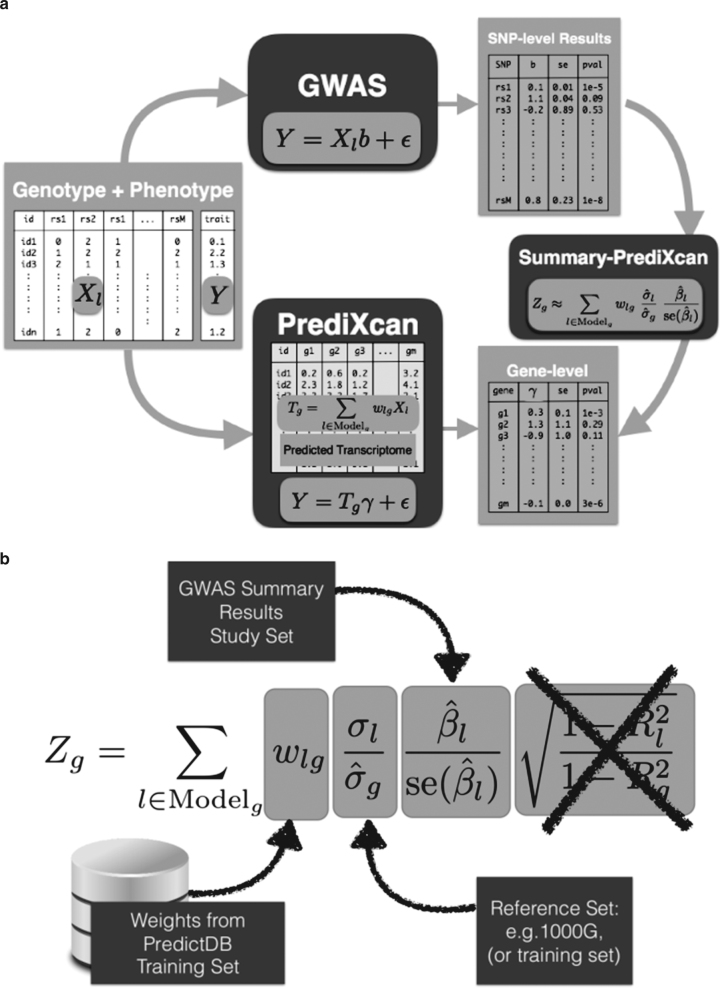
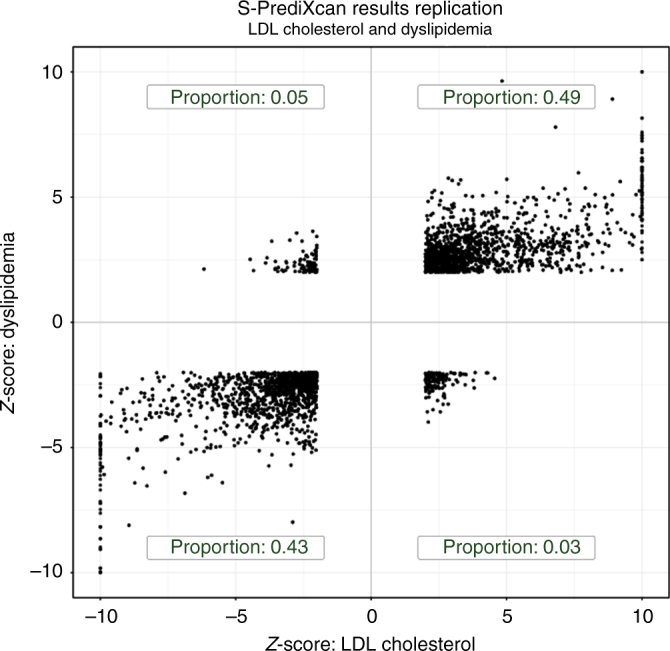
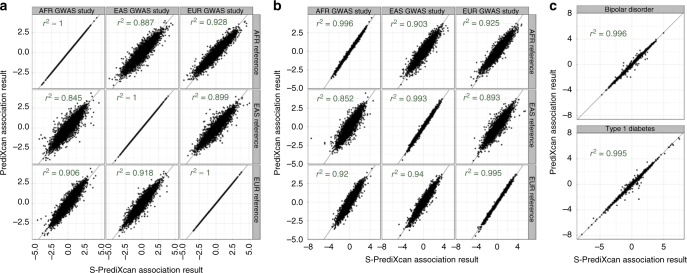
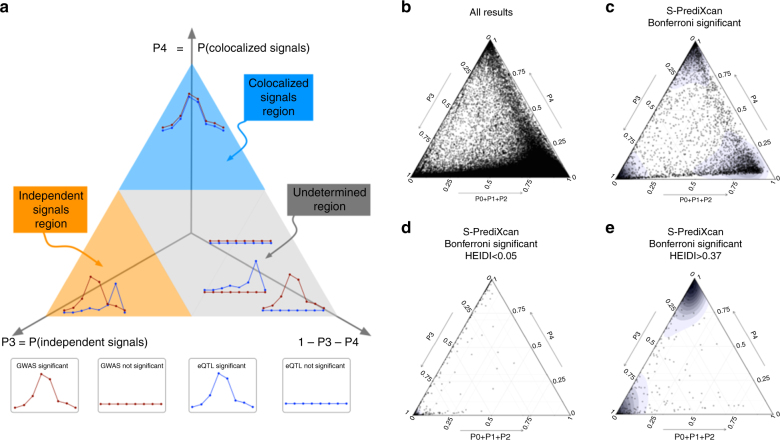
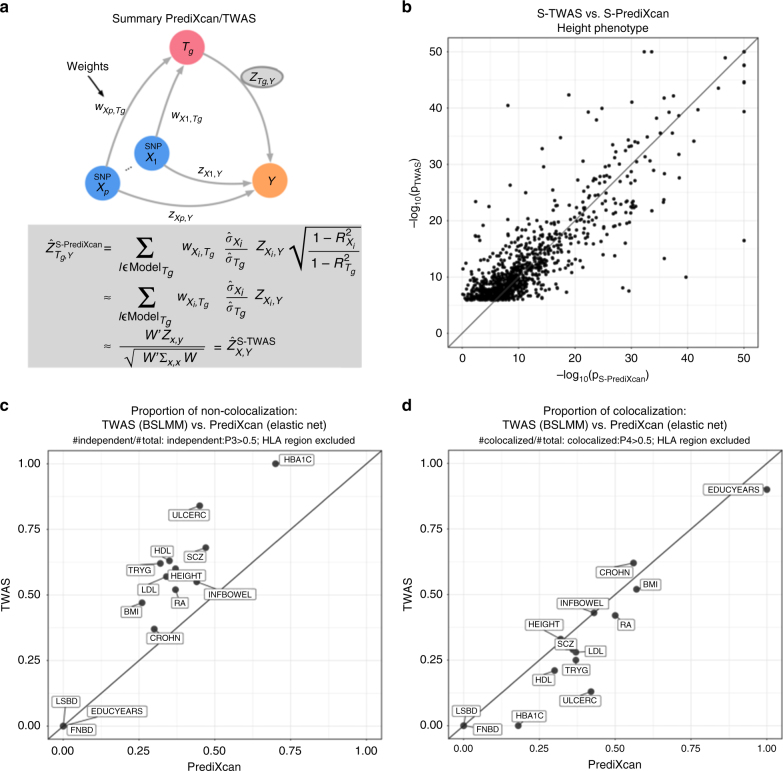
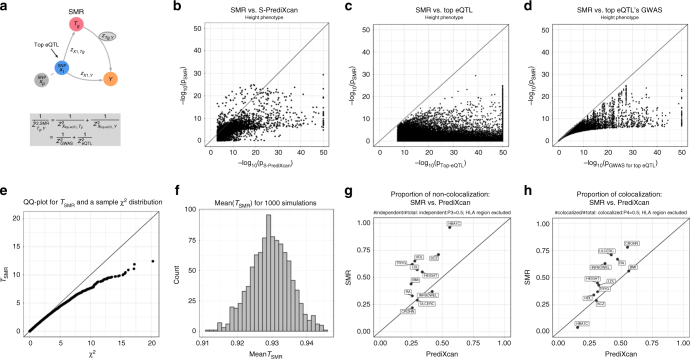
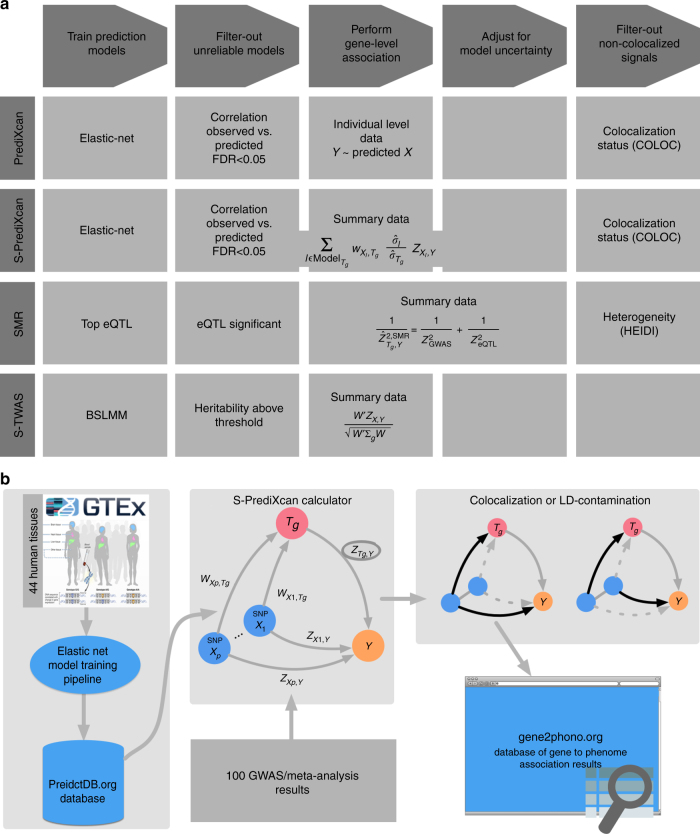
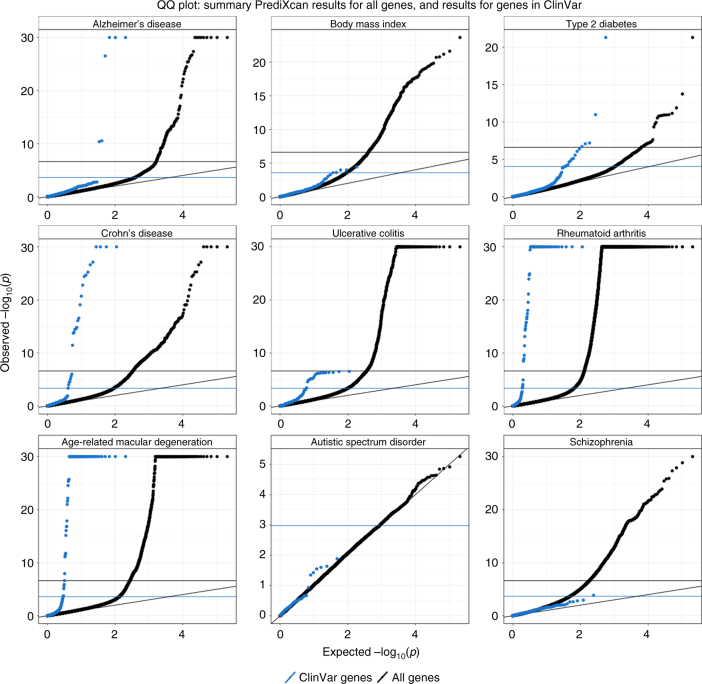
Scalable, integrative methods to understand mechanisms that link genetic variants with phenotypes are needed. Here we derive a mathematical expression to compute PrediXcan (a gene mapping approach) results using summary data (S-PrediXcan) and show its accuracy and general robustness to misspecified reference sets. We apply this framework to 44 GTEx tissues and 100+ phenotypes from GWAS and meta-analysis studies, creating a growing public catalog of associations that seeks to capture the effects of gene expression variation on human phenotypes. Replication in an independent cohort is shown. Most of the associations are tissue specific, suggesting context specificity of the trait etiology. Colocalized significant associations in unexpected tissues underscore the need for an agnostic scanning of multiple contexts to improve our ability to detect causal regulatory mechanisms. Monogenic disease genes are enriched among significant associations for related traits, suggesting that smaller alterations of these genes may cause a spectrum of milder phenotypes.
需要可扩展的、综合的方法来理解将遗传变异与表型联系起来的机制。在这里,我们推导出一个数学表达式,用于使用汇总数据(S-PrediXcan)计算 PrediXcan(一种基因映射方法)的结果,并展示其对指定参考集的准确性和普遍稳健性。我们将该框架应用于 44 个 GTEx 组织和来自 GWAS 和荟萃分析研究的 100 多个表型,创建了一个不断增长的公共关联目录,旨在捕捉基因表达变异对人类表型的影响。在一个独立的队列中进行了复制。大多数关联是组织特异性的,这表明性状病因具有特定的背景特异性。在意想不到的组织中发现的共定位显著关联强调需要对多个背景进行无偏扫描,以提高我们检测因果调节机制的能力。与相关性状相关的显著关联中富集了单基因疾病基因,这表明这些基因的较小改变可能导致一系列更轻微的表型。