Department of Pharmacology and Pharmacotherapy, Medical School, University of Pécs, Szigeti út 12, 7624 Pécs, Hungary.
Chemistry Doctoral School, University of Szeged, Dugonics tér 13, 6720 Szeged, Hungary.
Int J Mol Sci. 2019 Jan 19;20(2):422. doi: 10.3390/ijms20020422.
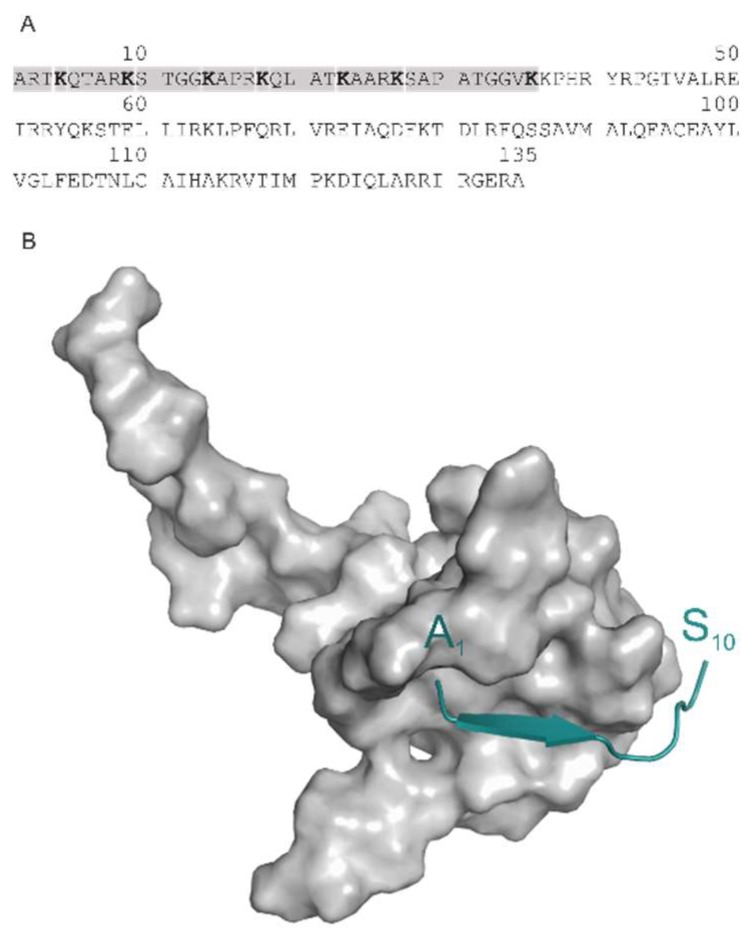
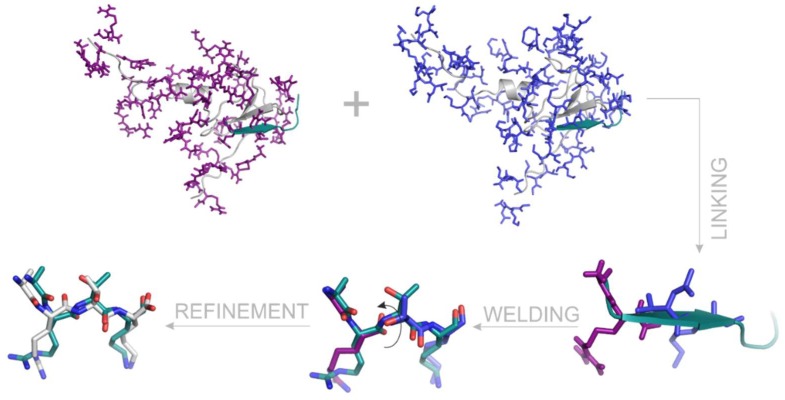
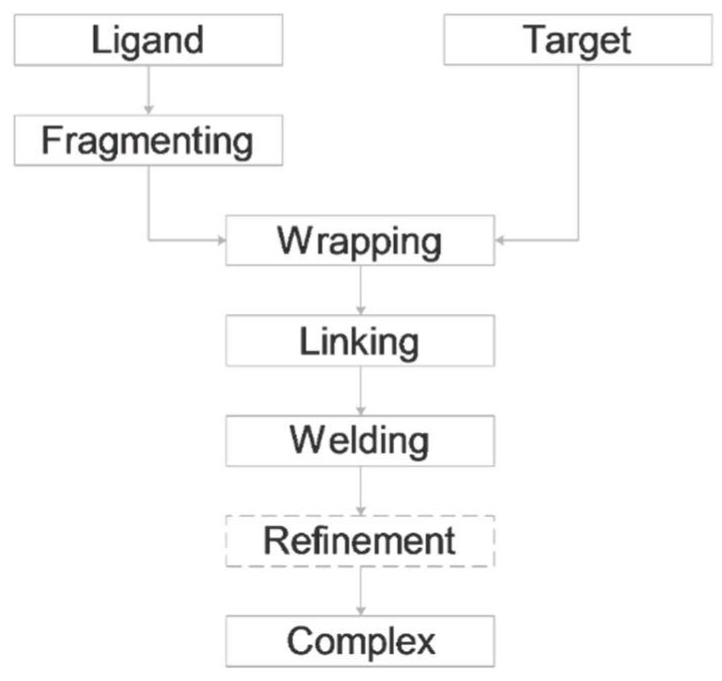
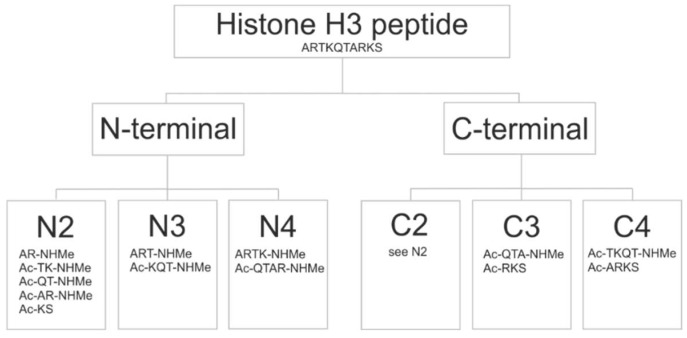
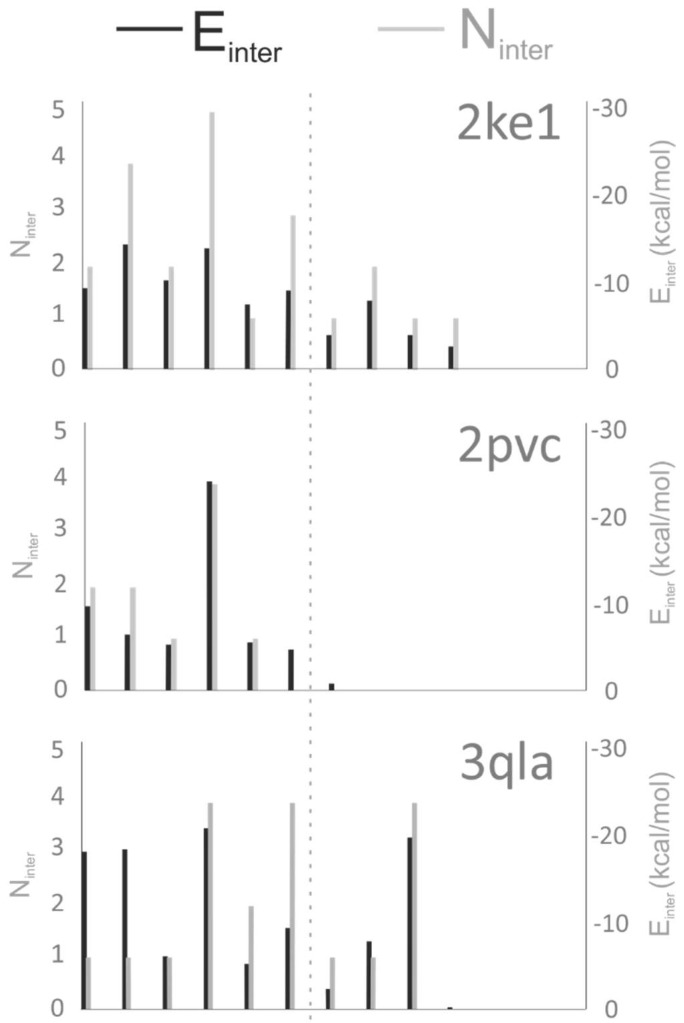
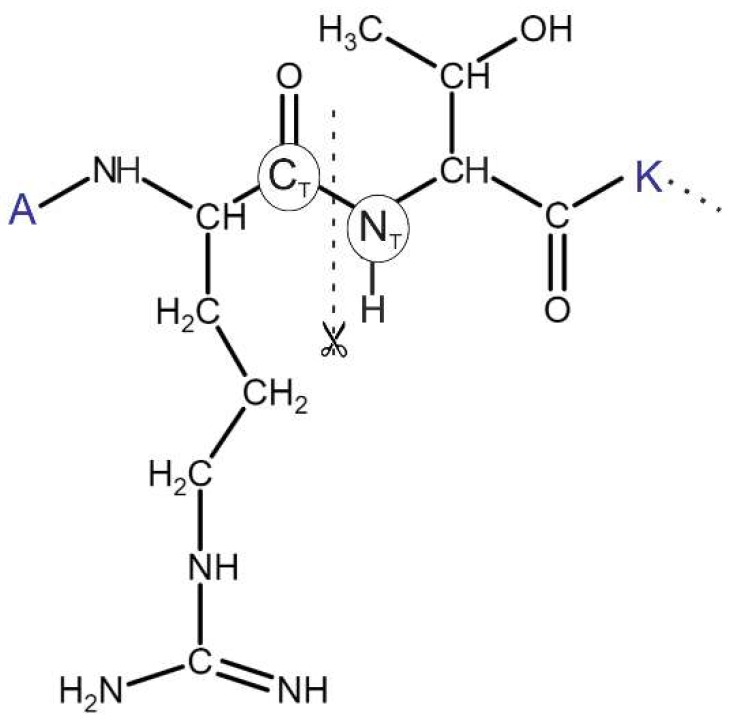
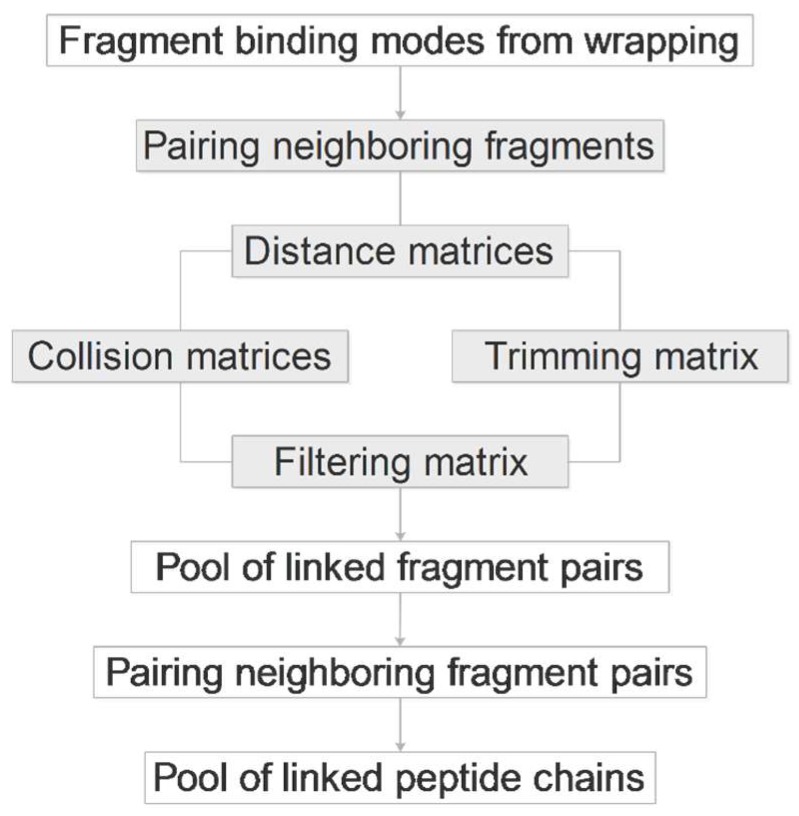
Histones serve as protein spools for winding the DNA in the nucleosome. High variability of their post-translational modifications result in a unique code system often responsible for the pathomechanisms of epigenetics-based diseases. Decoding is performed by reader proteins via complex formation with the N-terminal peptide tails of histones. Determination of structures of histone-reader complexes would be a key to unravel the histone code and the design of new drugs. However, the large number of possible histone complex variations imposes a true challenge for experimental structure determination techniques. Calculation of such complexes is difficult due to considerable size and flexibility of peptides and the shallow binding surfaces of the readers. Moreover, location of the binding sites is often unknown, which requires a blind docking search over the entire surface of the target protein. To accelerate the work in this field, a new approach is presented for prediction of the structure of histone H3 peptide tails docked to their targets. Using a fragmenting protocol and a systematic blind docking method, a collection of well-positioned fragments of the H3 peptide is produced. After linking the fragments, reconstitution of anchoring regions of the target-bound H3 peptide conformations was possible. As a first attempt of combination of blind and fragment docking approaches, our new method is named fragment blind docking (FBD).
组蛋白作为蛋白质线轴,用于将 DNA 缠绕在核小体中。它们的翻译后修饰的高度可变性导致了独特的编码系统,通常负责基于表观遗传学的疾病的发病机制。通过与组蛋白 N 端肽尾形成复合物,阅读器蛋白对其进行解码。确定组蛋白-阅读器复合物的结构将是揭示组蛋白密码和设计新药的关键。然而,大量可能的组蛋白复合物变体给实验结构确定技术带来了真正的挑战。由于肽的相当大的尺寸和灵活性以及阅读器的浅结合表面,计算这样的复合物是困难的。此外,结合位点的位置通常是未知的,这需要在目标蛋白的整个表面上进行盲目对接搜索。为了加速该领域的工作,提出了一种新的方法来预测与靶标结合的组蛋白 H3 肽尾的结构。使用碎片协议和系统的盲目对接方法,产生了一组定位良好的 H3 肽片段。在连接片段后,可能重建与靶标结合的 H3 肽的锚固区域的构象。作为盲目和片段对接方法的首次尝试,我们的新方法被命名为片段盲目对接(FBD)。