Nekrasov Evgeny D, Kiselev Sergey L
Vavilov Institute of General Genetics Russian Academy of Science, Moscow, 119991, Russia.
J Stem Cells Regen Med. 2018 Dec 30;14(2):80-85. doi: 10.46582/jsrm.1402012. eCollection 2018.
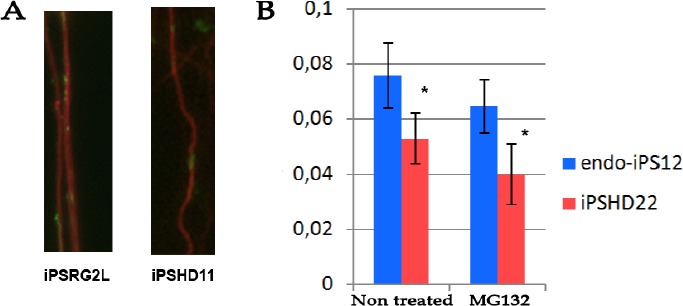
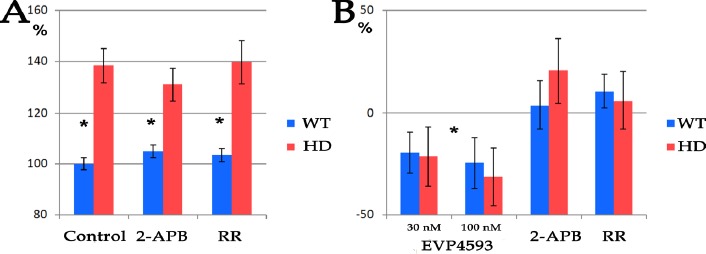
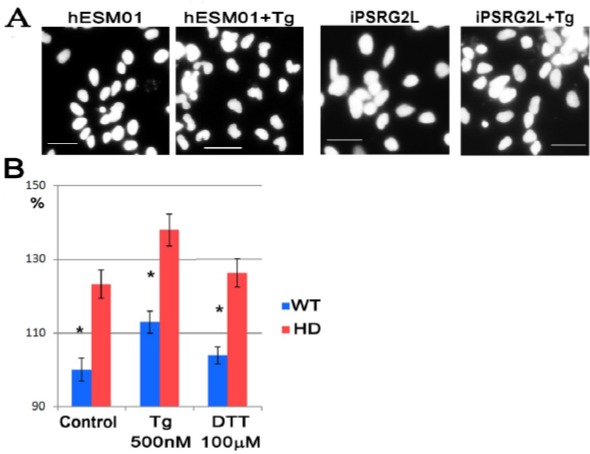
Huntington's disease (HD) is an inherited disease caused by an expansion of cytosine-adenine-guanine (CAG) repeats in the huntingtin gene () that ultimately leads to neurodegeneration. To study the molecular basis of this disease, induced pluripotent stem cells (iPSCs) generated from patients' fibroblasts were used to investigate axonal mitochondrial trafficking and the nature of nuclear indentations. Pathological and control iPSCs generated from patients with a low number of repeats were differentiated in striatal neurons of the brain. Mitochondrial density was measured along the axon using tubulin beta 3 co-staining in pathological and control neurons. To investigate the connection of nuclear roundness with calcium dysregulation, several calcium inhibitors were used. Proteasome system inhibition was applied to mimic premature neuronal ageing. We found that the mitochondrial density was approximately 7.6 ± 0.2 in neurites in control neurons but was only 5.3 ± 0.2 in mutant neurons with 40-44 CAG repeats (p-value <0.005). Neuronal ageing induced by proteasome inhibitor MG132 significantly decreased the mitochondrial density by 15% and 25% in control and mutant neurons to 6.5 ± 0.1 (p-value < 0.005) and 4.0 ± 0.3 (p-value < 0.005), respectively. Thus, for the first time, an impairment of mitochondrial trafficking in pathological neurons with endogenous mutant huntingtin was demonstrated. We found that inhibiting the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA), the ryanodine-receptor (RyR) or the inositol 1,4,5-trisphosphate receptor (IP3R) by specific inhibitors did not specifically affect the nuclear roundness or survival of pathological neurons differentiated from patient iPSCs. Therefore, nuclear calcium homeostasis is not directly associated with HD pathology. Identifying HD iPSCs and differentiating from them neurons provide a unique system for modelling the disease . Impairments of mitochondrial trafficking and nuclear roundness manifest long before the disease onset, while premature neuronal ageing enhances differences in mitochondrial distribution.
亨廷顿舞蹈症(HD)是一种遗传性疾病,由亨廷顿基因()中胞嘧啶 - 腺嘌呤 - 鸟嘌呤(CAG)重复序列的扩增引起,最终导致神经退行性变。为了研究这种疾病的分子基础,利用患者成纤维细胞生成的诱导多能干细胞(iPSC)来研究轴突线粒体运输以及核凹陷的性质。将来自重复次数较少的患者的病理iPSC和对照iPSC分化为脑内纹状体神经元。在病理和对照神经元中,使用微管蛋白β3共染色沿轴突测量线粒体密度。为了研究核圆度与钙调节异常之间的联系,使用了几种钙抑制剂。应用蛋白酶体系统抑制来模拟神经元过早衰老。我们发现,对照神经元神经突中的线粒体密度约为7.6±0.2,但在具有40 - 44个CAG重复序列的突变神经元中仅为5.3±0.2(p值<0.005)。蛋白酶体抑制剂MG132诱导的神经元衰老在对照和突变神经元中分别使线粒体密度显著降低15%和25%,降至6.5±0.1(p值<0.005)和4.0±0.3(p值<0.005)。因此,首次证明了内源性突变亨廷顿蛋白的病理神经元中线粒体运输受损。我们发现,用特异性抑制剂抑制肌浆网/内质网Ca2 + - ATP酶(SERCA)、兰尼碱受体(RyR)或肌醇1,4,5 - 三磷酸受体(IP3R)不会特异性影响从患者iPSC分化而来的病理神经元的核圆度或存活。因此,核钙稳态与HD病理没有直接关联。鉴定HD iPSC并从中分化出神经元为该疾病建模提供了一个独特的系统。线粒体运输和核圆度的损害在疾病发作前很久就已显现,而过早的神经元衰老加剧了线粒体分布的差异。