Nekrasov Evgeny D, Vigont Vladimir A, Klyushnikov Sergey A, Lebedeva Olga S, Vassina Ekaterina M, Bogomazova Alexandra N, Chestkov Ilya V, Semashko Tatiana A, Kiseleva Elena, Suldina Lyubov A, Bobrovsky Pavel A, Zimina Olga A, Ryazantseva Maria A, Skopin Anton Yu, Illarioshkin Sergey N, Kaznacheyeva Elena V, Lagarkova Maria A, Kiselev Sergey L
Vavilov Institute of General Genetics, Russian Academy of Sciences, Moscow, 119333, Russia.
Institute of Cytology, Russian Academy of Sciences, St. Petersburg, 194064, Russia.
Mol Neurodegener. 2016 Apr 14;11:27. doi: 10.1186/s13024-016-0092-5.
Huntington's disease (HD) is an incurable hereditary neurodegenerative disorder, which manifests itself as a loss of GABAergic medium spiny (GABA MS) neurons in the striatum and caused by an expansion of the CAG repeat in exon 1 of the huntingtin gene. There is no cure for HD, existing pharmaceutical can only relieve its symptoms.
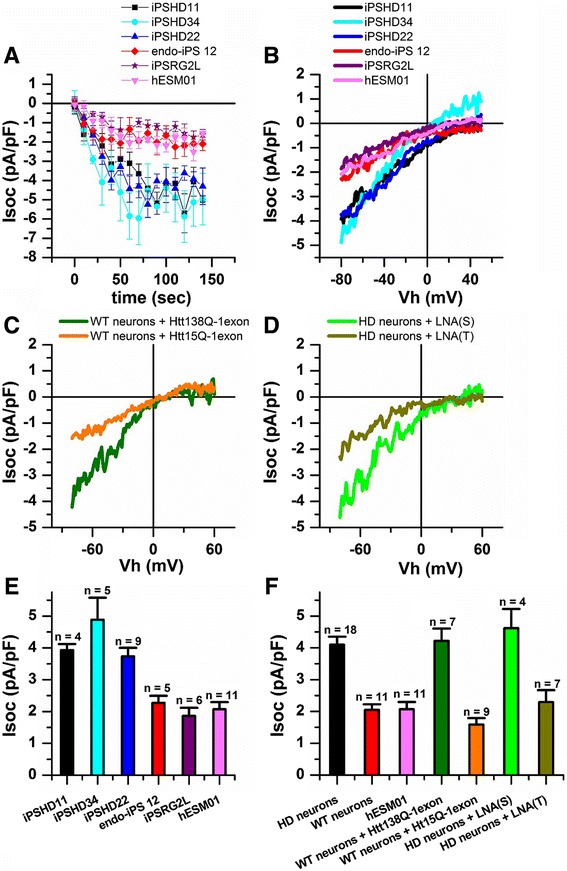
Here, induced pluripotent stem cells were established from patients with low CAG repeat expansion in the huntingtin gene, and were then efficiently differentiated into GABA MS-like neurons (GMSLNs) under defined culture conditions. The generated HD GMSLNs recapitulated disease pathology in vitro, as evidenced by mutant huntingtin protein aggregation, increased number of lysosomes/autophagosomes, nuclear indentations, and enhanced neuronal death during cell aging. Moreover, store-operated channel (SOC) currents were detected in the differentiated neurons, and enhanced calcium entry was reproducibly demonstrated in all HD GMSLNs genotypes. Additionally, the quinazoline derivative, EVP4593, reduced the number of lysosomes/autophagosomes and SOC currents in HD GMSLNs and exerted neuroprotective effects during cell aging.
Our data is the first to demonstrate the direct link of nuclear morphology and SOC calcium deregulation to mutant huntingtin protein expression in iPSCs-derived neurons with disease-mimetic hallmarks, providing a valuable tool for identification of candidate anti-HD drugs. Our experiments demonstrated that EVP4593 may be a promising anti-HD drug.
亨廷顿舞蹈症(HD)是一种无法治愈的遗传性神经退行性疾病,表现为纹状体中γ-氨基丁酸能中等棘状(GABA MS)神经元的丧失,由亨廷顿基因外显子1中CAG重复序列的扩增引起。HD无法治愈,现有药物只能缓解其症状。
在此,我们从亨廷顿基因中CAG重复序列扩增较低的患者中建立了诱导多能干细胞,然后在特定培养条件下将其高效分化为GABA MS样神经元(GMSLNs)。所生成的HD GMSLNs在体外重现了疾病病理,表现为突变型亨廷顿蛋白聚集、溶酶体/自噬体数量增加、核凹陷以及细胞衰老过程中神经元死亡增加。此外,在分化的神经元中检测到了储存性钙通道(SOC)电流,并且在所有HD GMSLNs基因型中均再现了增强的钙内流。此外,喹唑啉衍生物EVP4593减少了HD GMSLNs中的溶酶体/自噬体数量和SOC电流,并在细胞衰老过程中发挥了神经保护作用。
我们的数据首次证明了核形态和SOC钙调节异常与具有疾病模拟特征的诱导多能干细胞衍生神经元中突变型亨廷顿蛋白表达之间的直接联系,为鉴定候选抗HD药物提供了有价值的工具。我们的实验表明EVP4593可能是一种有前景的抗HD药物。