School of Biochemistry and Immunology, Trinity College Institute of Neuroscience & Trinity Biomedical Sciences Institute, Trinity College Dublin, Dublin, Republic of Ireland.
Department of Medicine, Division of Pulmonary, Critical Care and Sleep Medicine, University of Florida, Gainesville, Florida.
Glia. 2019 Jul;67(7):1254-1276. doi: 10.1002/glia.23592. Epub 2019 Jan 25.
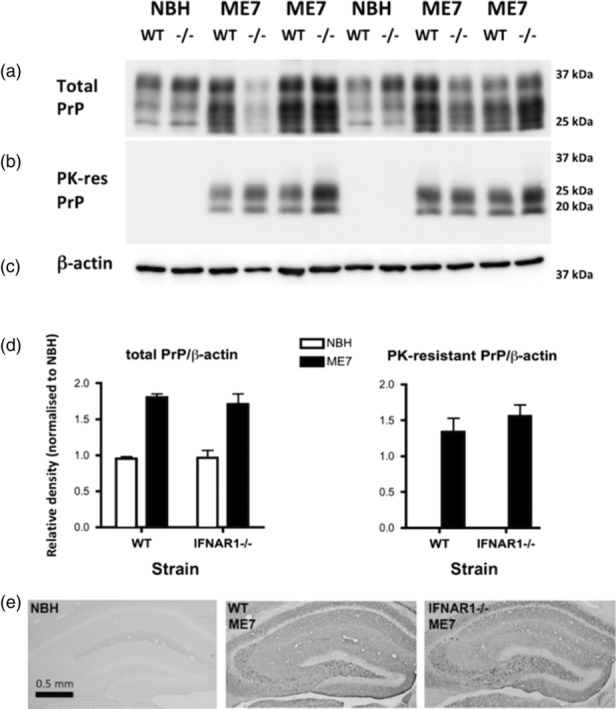
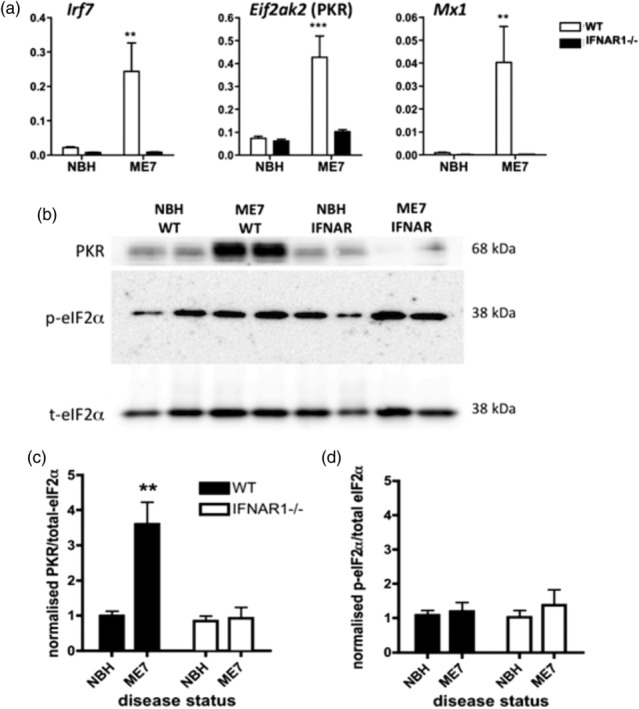
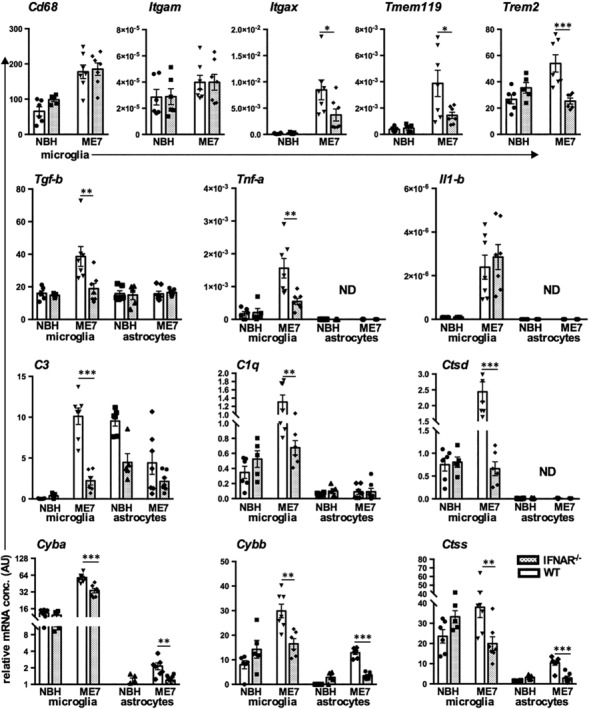
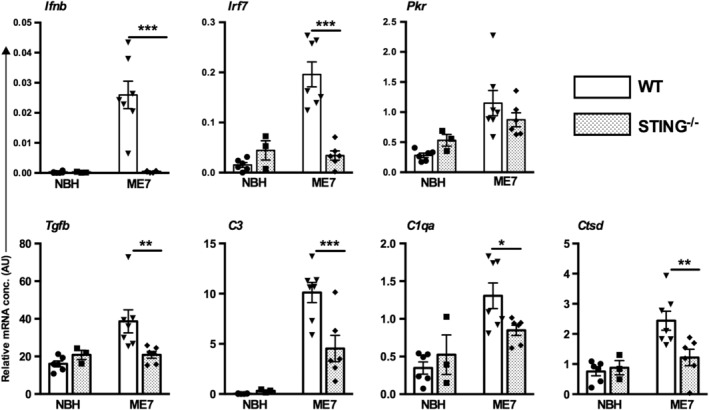
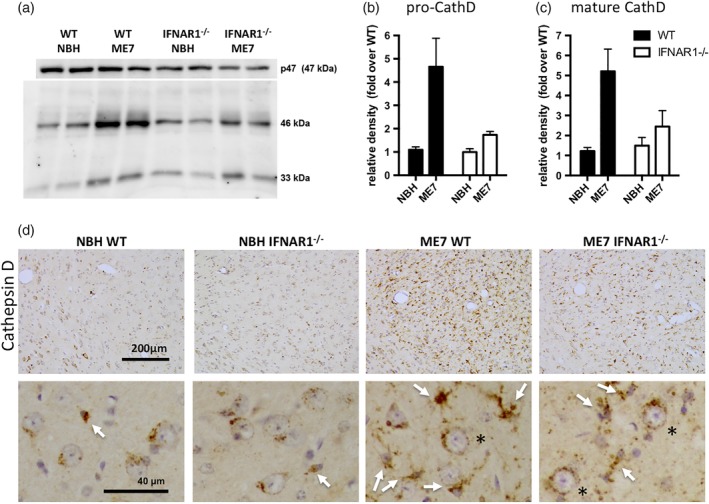
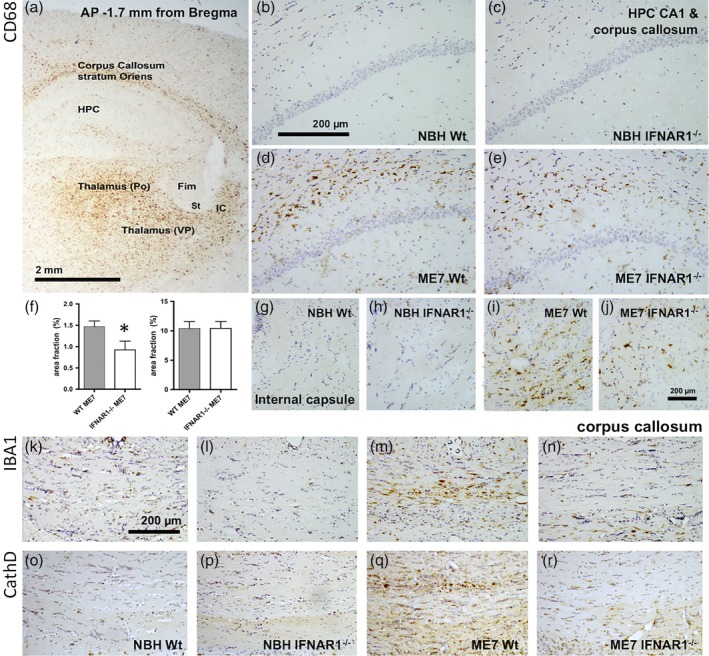
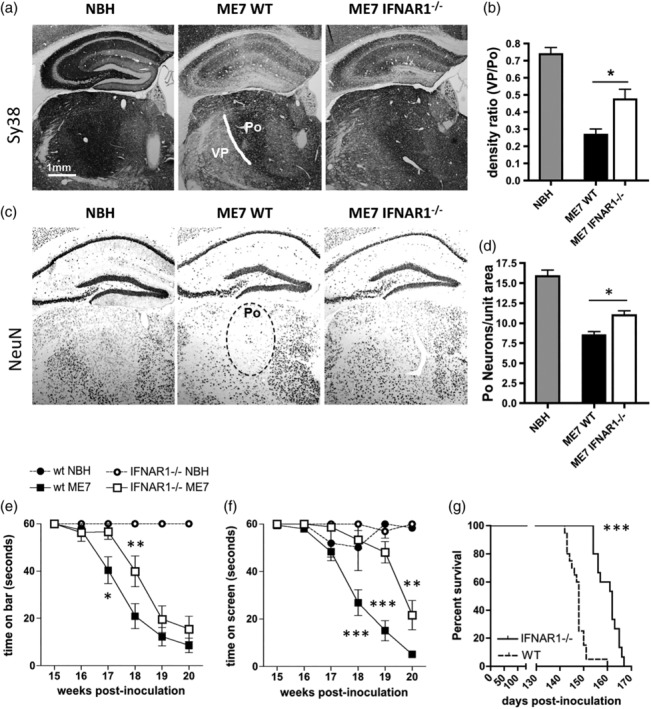
Type I interferons (IFN-I) are the principal antiviral molecules of the innate immune system and can be made by most cell types, including central nervous system cells. IFN-I has been implicated in neuroinflammation during neurodegeneration, but its mechanism of induction and its consequences remain unclear. In the current study, we assessed expression of IFN-I in murine prion disease (ME7) and examined the contribution of the IFN-I receptor IFNAR1 to disease progression. The data indicate a robust IFNβ response, specifically in microglia, with evidence of IFN-dependent genes in both microglia and astrocytes. This IFN-I response was absent in stimulator of interferon genes (STING ) mice. Microglia showed increased numbers and activated morphology independent of genotype, but transcriptional signatures indicated an IFNAR1-dependent neuroinflammatory phenotype. Isolation of microglia and astrocytes demonstrated disease-associated microglial induction of Tnfα, Tgfb1, and of phagolysosomal system transcripts including those for cathepsins, Cd68, C1qa, C3, and Trem2, which were diminished in IFNAR1 and STING deficient mice. Microglial increases in activated cathepsin D, and CD68 were significantly reduced in IFNAR1 mice, particularly in white matter, and increases in COX-1 expression, and prostaglandin synthesis were significantly mitigated. Disease progressed more slowly in IFNAR1 mice, with diminished synaptic and neuronal loss and delayed onset of neurological signs and death but without effect on proteinase K-resistant PrP levels. Therefore, STING-dependent IFN-I influences microglial phenotype and influences neurodegenerative progression despite occurring secondary to initial degenerative changes. These data expand our mechanistic understanding of IFN-I induction and its impact on microglial function during chronic neurodegeneration.
I 型干扰素 (IFN-I) 是先天免疫系统的主要抗病毒分子,可由包括中枢神经系统细胞在内的大多数细胞类型产生。IFN-I 已被牵连到神经退行性变中的神经炎症中,但诱导机制及其后果仍不清楚。在当前的研究中,我们评估了 IFN-I 在鼠朊病毒病 (ME7) 中的表达,并研究了 IFN-I 受体 IFNAR1 对疾病进展的贡献。数据表明存在强烈的 IFNβ 反应,特别是在小胶质细胞中,并且在小胶质细胞和星形胶质细胞中都有 IFN 依赖性基因的证据。这种 IFN-I 反应在干扰素基因刺激因子 (STING) 小鼠中不存在。小胶质细胞的数量增加且形态激活,但转录特征表明存在依赖 IFNAR1 的神经炎症表型。小胶质细胞和星形胶质细胞的分离表明,疾病相关的小胶质细胞诱导了 TNFα、Tgfb1 和吞噬溶酶体系统转录物,包括组织蛋白酶、CD68、C1qa、C3 和 Trem2 的转录物,这些在 IFNAR1 和 STING 缺陷型小鼠中减少。IFNAR1 小鼠中疾病相关的小胶质细胞激活的组织蛋白酶 D 和 CD68 显著减少,特别是在白质中,COX-1 表达和前列腺素合成的增加显著减轻。IFNAR1 小鼠的疾病进展更缓慢,突触和神经元丢失减少,神经症状和死亡的发作延迟,但对蛋白酶 K 抗性 PrP 水平没有影响。因此,STING 依赖性 IFN-I 影响小胶质细胞表型,并影响神经退行性进展,尽管发生在初始退行性变化之后。这些数据扩展了我们对 IFN-I 诱导及其在慢性神经退行性变中小胶质细胞功能中的作用的机制理解。