Human Genetics, Wellcome Sanger Institute, Hinxton, UK.
European Molecular Biology Laboratory, European Bioinformatics Institute (EMBL-EBI), Hinxton, UK.
Nat Genet. 2019 Feb;51(2):343-353. doi: 10.1038/s41588-018-0322-6. Epub 2019 Jan 28.
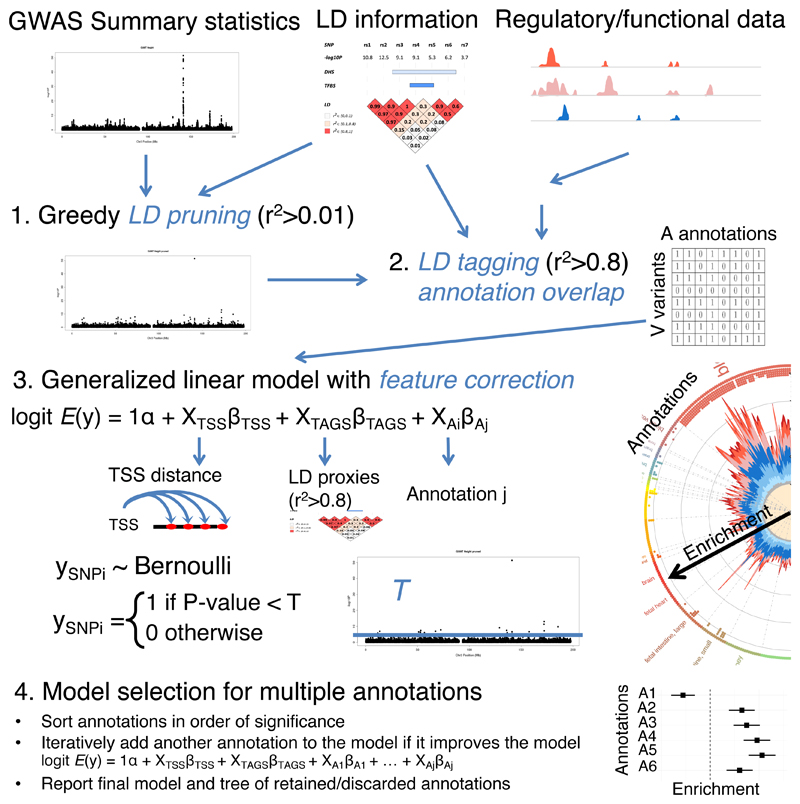
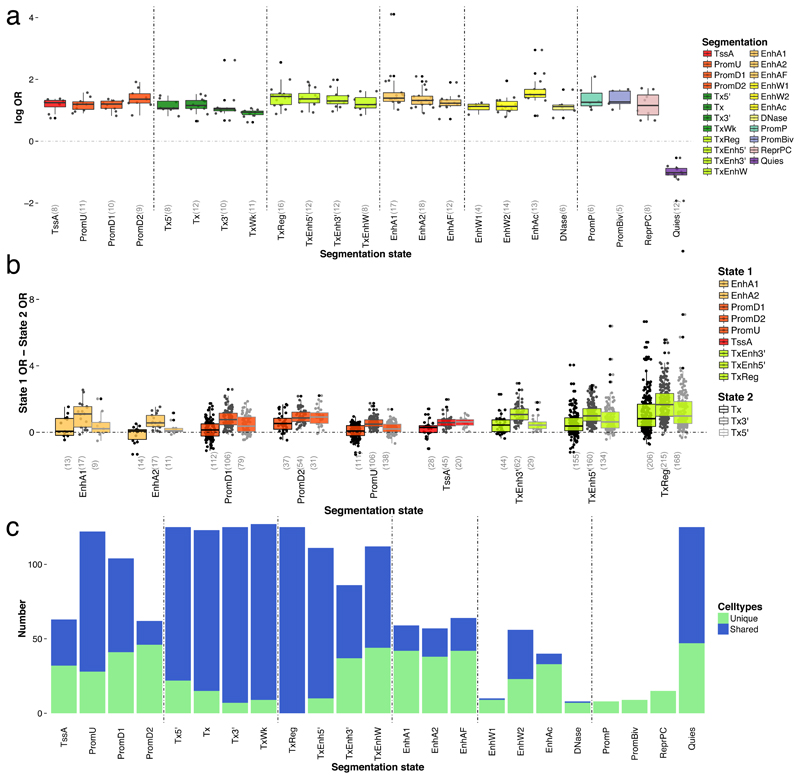
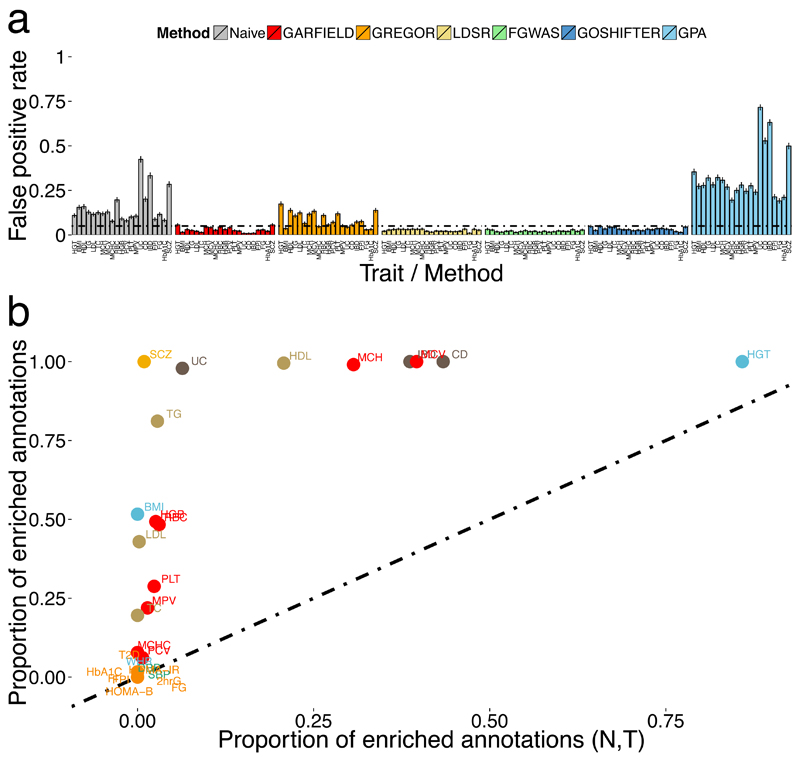
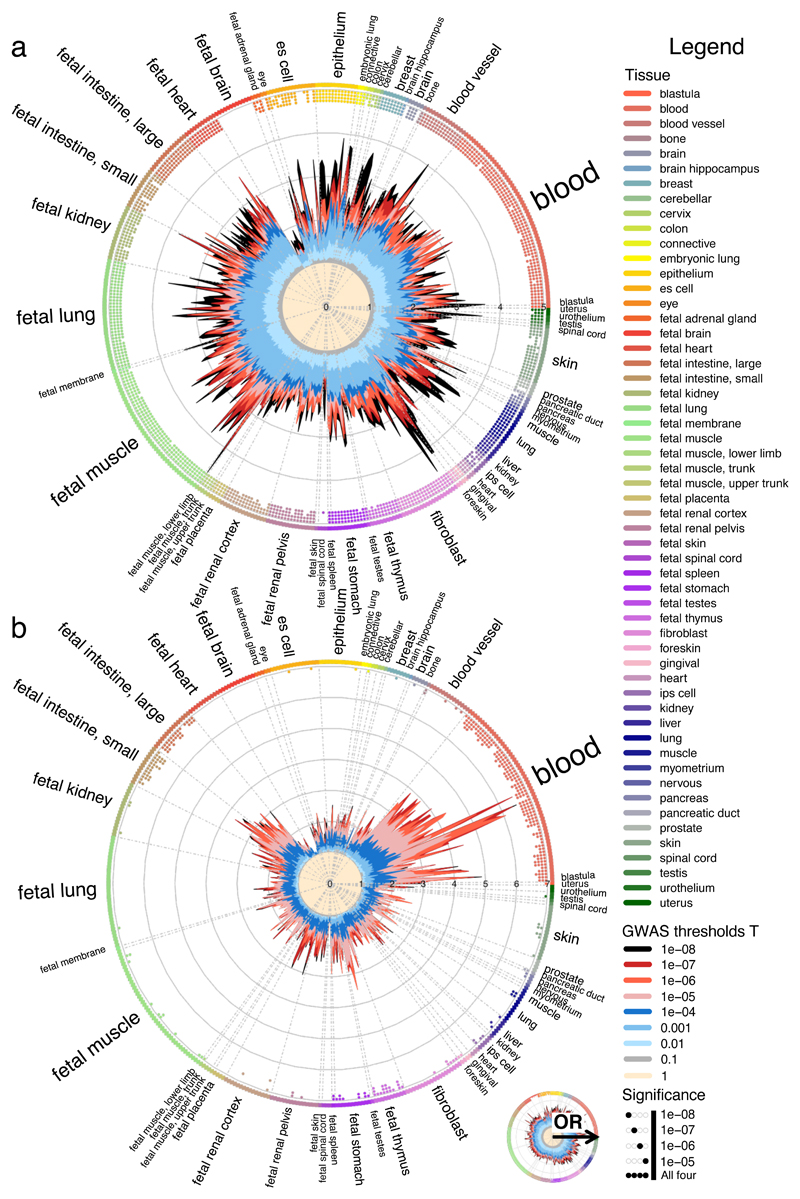
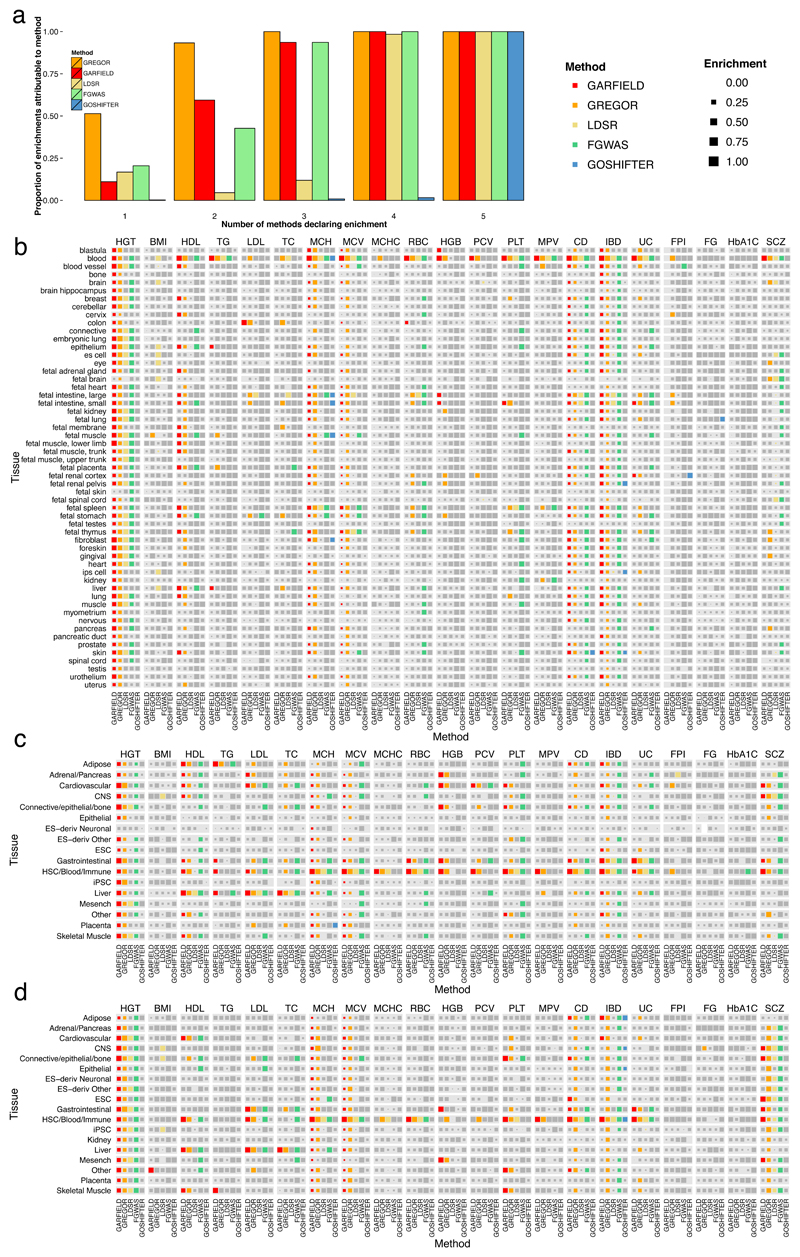
Loci discovered by genome-wide association studies predominantly map outside protein-coding genes. The interpretation of the functional consequences of non-coding variants can be greatly enhanced by catalogs of regulatory genomic regions in cell lines and primary tissues. However, robust and readily applicable methods are still lacking by which to systematically evaluate the contribution of these regions to genetic variation implicated in diseases or quantitative traits. Here we propose a novel approach that leverages genome-wide association studies' findings with regulatory or functional annotations to classify features relevant to a phenotype of interest. Within our framework, we account for major sources of confounding not offered by current methods. We further assess enrichment of genome-wide association studies for 19 traits within Encyclopedia of DNA Elements- and Roadmap-derived regulatory regions. We characterize unique enrichment patterns for traits and annotations driving novel biological insights. The method is implemented in standalone software and an R package, to facilitate its application by the research community.
全基因组关联研究发现的基因座主要位于蛋白质编码基因之外。细胞系和原代组织中调控基因组区域的目录可以极大地增强对非编码变异功能后果的解释。然而,仍然缺乏稳健且易于应用的方法,无法系统地评估这些区域对疾病或数量性状中涉及的遗传变异的贡献。在这里,我们提出了一种新的方法,利用全基因组关联研究的结果与调控或功能注释来对与感兴趣的表型相关的特征进行分类。在我们的框架内,我们考虑了当前方法没有提供的主要混杂来源。我们进一步评估了全基因组关联研究在 DNA 元素百科全书和路线图衍生调控区域内 19 个特征上的富集情况。我们对特征和注释的独特富集模式进行了描述,这些模式为新的生物学见解提供了驱动力。该方法以独立软件和 R 包的形式实现,便于研究界应用。