Tufton Nicola, Shapiro Lucy, Sahdev Anju, Kumar Ajith V, Martin Lee, Drake William M, Akker Scott A, Storr Helen L
Department of Endocrinology, St Bartholomew's Hospital, Barts Health NHS Trust, London, UK.
Centre for Endocrinology, Barts and the London School of Medicine and Dentistry, Queen Mary University of London, London, UK.
Endocr Connect. 2019 Mar 1;8(3):162-172. doi: 10.1530/EC-18-0522.
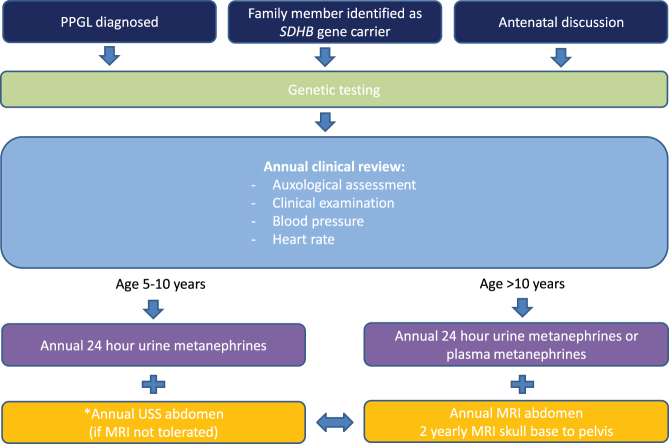
Objective Phaeochromocytomas (PCC) and paragangliomas (PGL) are rare in children. A large proportion of these are now understood to be due to underlying germline mutations. Here we focus on succinate dehydrogenase subunit B (SDHB) gene mutation carriers as these tumours carry a high risk of malignant transformation. There remains no current consensus with respect to optimal surveillance for asymptomatic carriers and those in whom the presenting tumour has been resected. Method We undertook a retrospective analysis of longitudinal clinical data of all children and adolescents with SDHB mutations followed up in a single UK tertiary referral centre. This included index cases that pre-dated the introduction of surveillance screening and asymptomatic carriers identified through cascade genetic testing. We also conducted a literature review to inform a suggested surveillance protocol for children and adolescents harbouring SDHB mutations. Results Clinical outcomes of a total of 38 children are presented: 8 index cases and 30 mutation-positive asymptomatic carriers with 175 patient years of follow-up data. Three of the eight index cases developed metachronous disease and two developed metastatic disease. Of the 30 asymptomatic carriers, 3 were found to have PGLs on surveillance screening. Conclusions Surveillance screening was well tolerated in our paediatric cohort and asymptomatic paediatric subjects. Screening can identify tumours before they become secretory and/or symptomatic, thereby facilitating surgical resection and reducing the chance of distant spread. We propose a regular screening protocol commencing at age 5 years in this at-risk cohort of patients.
目的 嗜铬细胞瘤(PCC)和副神经节瘤(PGL)在儿童中较为罕见。现在已知其中很大一部分是由潜在的种系突变引起的。在此,我们重点关注琥珀酸脱氢酶亚基B(SDHB)基因突变携带者,因为这些肿瘤具有很高的恶性转化风险。对于无症状携带者以及已切除首发肿瘤的患者,目前尚无关于最佳监测方案的共识。方法 我们对在英国一家三级转诊中心接受随访的所有携带SDHB突变的儿童和青少年的纵向临床数据进行了回顾性分析。这包括在监测筛查引入之前的索引病例以及通过级联基因检测确定的无症状携带者。我们还进行了文献综述,以制定针对携带SDHB突变的儿童和青少年的建议监测方案。结果 共呈现了38名儿童的临床结果:8例索引病例和30例突变阳性无症状携带者,有175患者年的随访数据。8例索引病例中有3例发生了异时性疾病,2例发生了转移性疾病。在30例无症状携带者中,3例在监测筛查中被发现患有PGL。结论 在我们的儿科队列和无症状儿科受试者中,监测筛查耐受性良好。筛查可以在肿瘤分泌和/或出现症状之前识别肿瘤,从而便于手术切除并减少远处转移的机会。我们建议在这个高危患者队列中从5岁开始采用定期筛查方案。