Department of Biochemistry and Molecular Biology, McGovern Medical School at Houston, University of Texas, Houston, Texas, USA.
Department of Nephrology, The First Xiangya Hospital of Central South University, Changsha, Hunan, PRC.
Am J Hypertens. 2019 Apr 22;32(5):476-485. doi: 10.1093/ajh/hpz018.
Although numerous recent studies have shown a strong link between inflammation and hypertension, the underlying mechanisms by which inflammatory cytokines induce hypertension remain to be fully elucidated. Hypertensive disorders are also associated with elevated pressor sensitivity. Tissue transglutaminase (TG2), a potent cross-linking enzyme, is known to be transcriptionally activated by inflammatory cytokines and stabilize angiotensin II (Ang II) receptor AT1 (AT1R) via ubiquitination-preventing posttranslational modification. Here we sought to investigate the TG2-mediated AT1R stabilization in inflammation-induced hypertension and its functional consequences with a focus on receptor abundance and Ang II responsiveness.
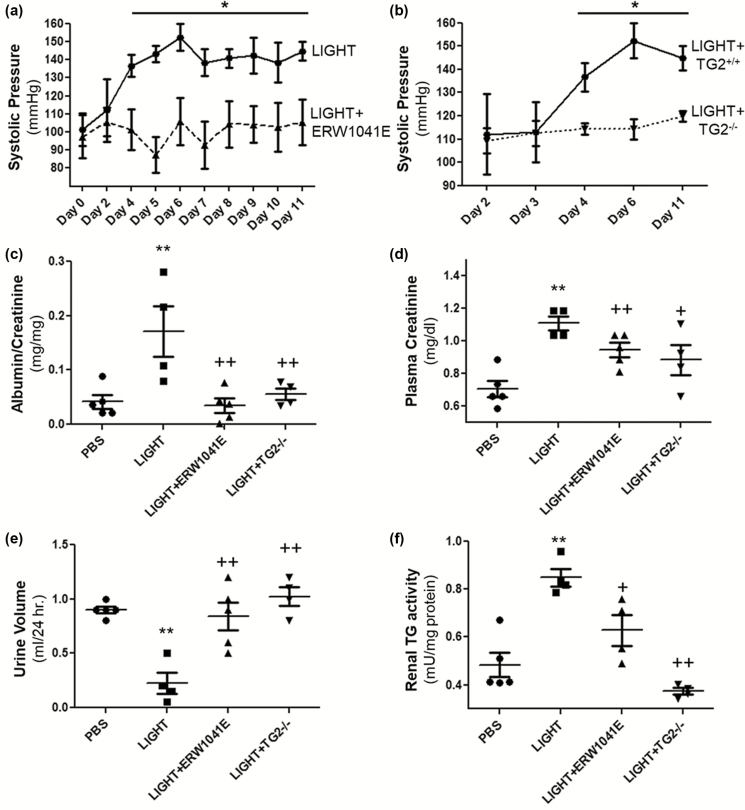
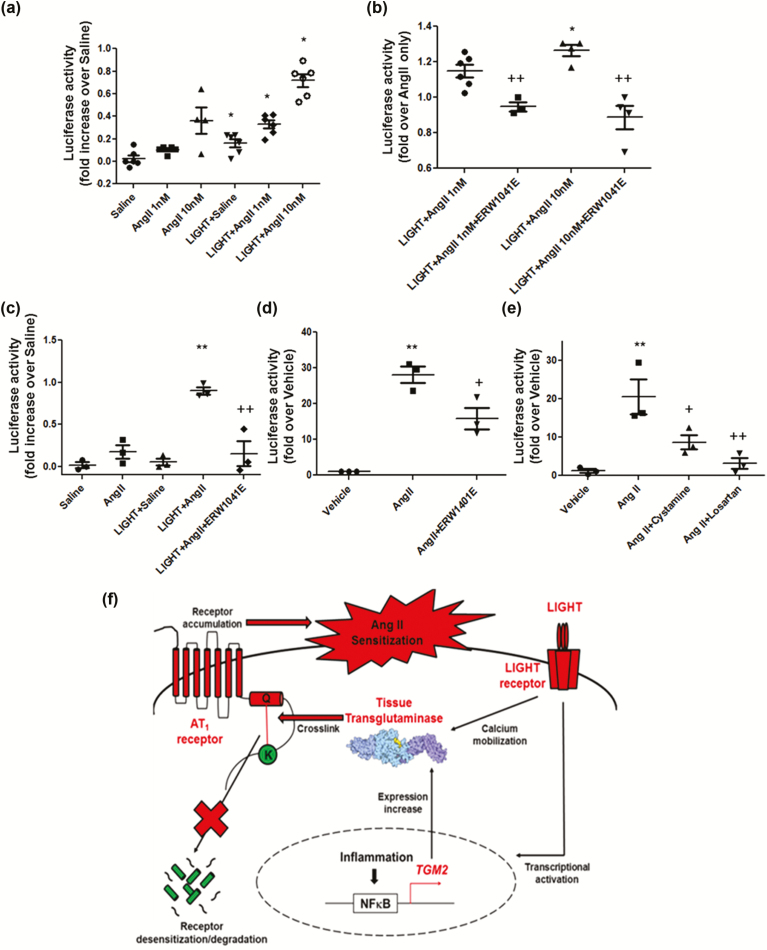
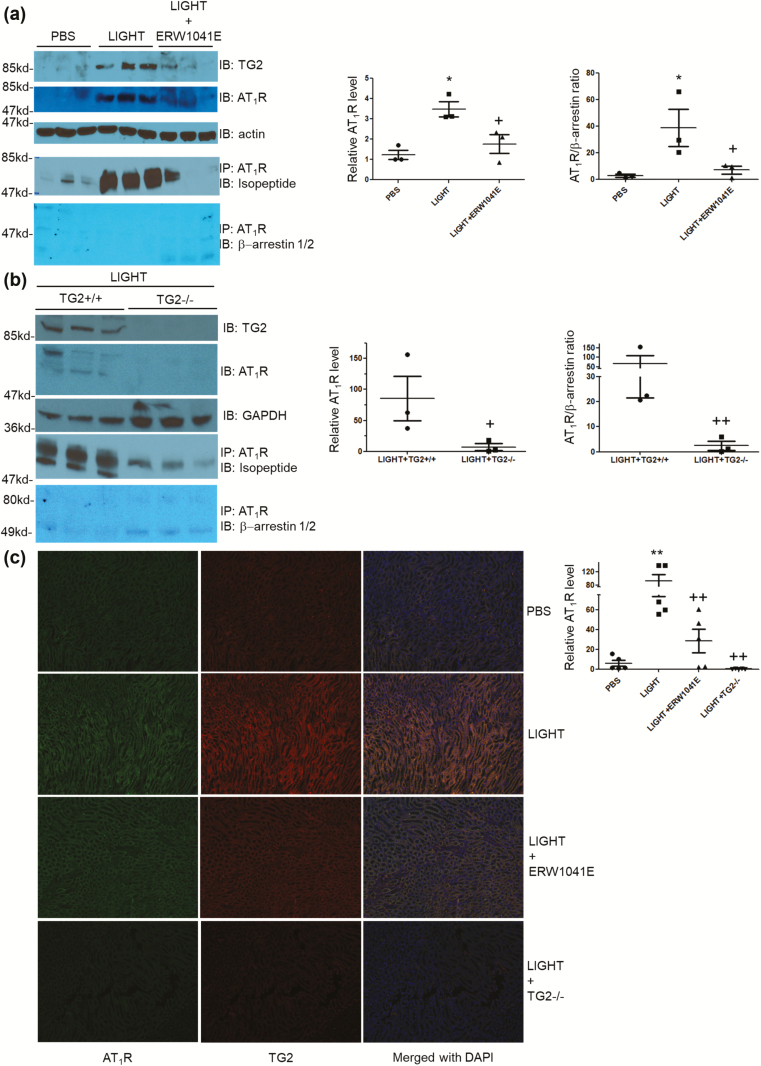
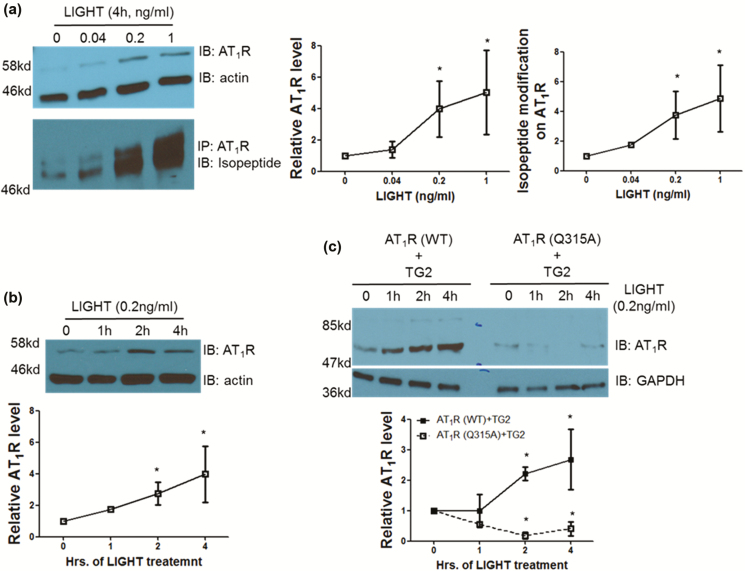
Using an experimental model of inflammation-induced hypertension established by introducing the pro-inflammatory tumor necrosis factor cytokine LIGHT, we provide pharmacologic and genetic evidence that TG2 is required for LIGHT-induced hypertension (systolic pressure on day 6: LIGHT = 152.3 ± 7.4 vs. LIGHT+ERW1041E [TG2 inhibitor] = 105.8 ± 13.1 or LIGHT+TG2-/- = 114.3 ± 4.3 mm Hg, P < 0.05, n = 4-5) and renal compromise (urine albumin/creatinine: LIGHT = 0.17 ± 0.05 vs. LIGHT+ERW1041E = 0.03 ± 0.01 or LIGHT+TG2-/- = 0.06 ± 0.01 mg/mg; plasma creatinine: LIGHT = 1.11 ± 0.04 vs. LIGHT+ERW1041E = 0.94 ± 0.04 or LIGHT+TG2-/- = 0.88 ± 0.09 mg/dl; urine volume: LIGHT = 0.23 ± 0.1 vs. LIGHT+ERW1041E = 0.84 ± 0.13 or LIGHT+TG2-/- = 1.02 ± 0.09 ml/24 hour on day 14, P < 0.05, n = 4-5). Our mechanistic studies showed that the TG2-mediated AT1R modification and accumulation (relative renal AT1R level: phosphate-buffered saline [PBS] = 1.23 ± 0.22, LIGHT = 3.49 ± 0.37, and LIGHT+ERW1041E = 1.77 ± 0.46, P < 0.05, n = 3; LIGHT+TG2+/+ = 85.28 ± 36.11 vs. LIGHT+TG2-/- = 7.01 ± 5.68, P < 0.05, n = 3) induced by LIGHT is associated with abrogated β-arrestin binding (AT1R/associated β-arrestin ratio: PBS = 2.62 ± 1.07, LIGHT = 38.60 ± 13.91, and LIGHT+ERW1041E = 6.97 ± 2.91, P < 0.05, n = 3; LIGHT+TG2+/+ = 66.43 ± 44.81 vs. LIGHT+TG2-/- = 2.45 ± 1.78, P < 0.01, n = 3) and could be found in renal medulla tubules of kidneys (relative tubular AT1R level: PBS = 5.91 ± 2.93, LIGHT = 92.82 ± 19.54, LIGHT+ERW1041E = 28.49 ± 11.65, and LIGHT+TG2-/- = 0.14 ± 0.10, P < 0.01, n = 5) and the blood vasculature (relative vascular AT1R level: PBS = 0.70 ± 0.30, LIGHT = 13.75 ± 2.49, and LIGHT+ERW1041E = 3.28 ± 0.87, P < 0.01, n = 3), 2 of the tissues highly related to the genesis of hypertension. Our in vitro cellular assays showed that LIGHT stimulation triggered a rapid TG2-dependent increase in the abundance of AT1Rs (relative AT1R level after 2-hour LIGHT treatment: AT1R (WT)+TG2 = 2.21 ± 0.23, AT1R (Q315A)+TG2 = 0.18 ± 0.23, P < 0.05 vs. starting point = 1, n = 2) and downstream calcium signaling (fold increase in NFAT-driven luciferase activity: Saline = 0.02 ± 0.03, Ang II = 0.17 ± 0.08, LIGHT = 0.05 ± 0.04, LIGHT+Ang II = 0.90 ± 0.04 (P < 0.01 vs. Ang II), and LIGHT+Ang II+ERW1041E = 0.15 ± 0.15 (P < 0.01 vs. LIGHT+Ang II), n = 3).
Our data indicate an essential and systemic role for TG2 in bridging inflammation to hypertension via its posttranslational modifications stabilizing AT1 receptor and sensitizing Ang II. Our findings also suggest that TG2 inhibitors could be used as a novel group of cardiovascular agents.
尽管最近有大量研究表明炎症与高血压之间存在很强的关联,但炎症细胞因子引起高血压的确切机制仍未完全阐明。高血压疾病也与血压升高的敏感性有关。组织转谷氨酰胺酶(TG2)是一种有效的交联酶,已知可被炎症细胞因子转录激活,并通过阻止翻译后修饰稳定血管紧张素 II(Ang II)受体 AT1(AT1R)。在这里,我们试图研究 TG2 在炎症诱导的高血压中的作用及其对受体丰度和 Ang II 反应性的影响。
我们使用了一种通过引入促炎肿瘤坏死因子细胞因子 LIGHT 建立的炎症诱导的高血压实验模型,提供了药理学和遗传学证据,表明 TG2 是 LIGHT 诱导的高血压(第 6 天收缩压:LIGHT = 152.3 ± 7.4 与 LIGHT+ERW1041E [TG2 抑制剂] = 105.8 ± 13.1 或 LIGHT+TG2-/- = 114.3 ± 4.3 mmHg,P < 0.05,n = 4-5)和肾脏损伤(尿白蛋白/肌酐:LIGHT = 0.17 ± 0.05 与 LIGHT+ERW1041E = 0.03 ± 0.01 或 LIGHT+TG2-/- = 0.06 ± 0.01 mg/mg;血浆肌酐:LIGHT = 1.11 ± 0.04 与 LIGHT+ERW1041E = 0.94 ± 0.04 或 LIGHT+TG2-/- = 0.88 ± 0.09 mg/dl;尿体积:LIGHT = 0.23 ± 0.1 与 LIGHT+ERW1041E = 0.84 ± 0.13 或 LIGHT+TG2-/- = 1.02 ± 0.09 ml/24 小时,P < 0.05,n = 4-5)所必需的。我们的机制研究表明,TG2 介导的 AT1R 修饰和积累(相对肾 AT1R 水平:磷酸盐缓冲盐水 [PBS] = 1.23 ± 0.22,LIGHT = 3.49 ± 0.37,和 LIGHT+ERW1041E = 1.77 ± 0.46,P < 0.05,n = 3;LIGHT+TG2+/+ = 85.28 ± 36.11 与 LIGHT+TG2-/- = 7.01 ± 5.68,P < 0.05,n = 3)与结合β-抑制蛋白的减少(AT1R/结合β-抑制蛋白比值:PBS = 2.62 ± 1.07,LIGHT = 38.60 ± 13.91,和 LIGHT+ERW1041E = 6.97 ± 2.91,P < 0.05,n = 3;LIGHT+TG2+/+ = 66.43 ± 44.81 与 LIGHT+TG2-/- = 2.45 ± 1.78,P < 0.01,n = 3)有关,并且可以在肾脏髓质肾小管中发现(相对肾小管 AT1R 水平:PBS = 5.91 ± 2.93,LIGHT = 92.82 ± 19.54,LIGHT+ERW1041E = 28.49 ± 11.65,和 LIGHT+TG2-/- = 0.14 ± 0.10,P < 0.01,n = 5)和血管(相对血管 AT1R 水平:PBS = 0.70 ± 0.30,LIGHT = 13.75 ± 2.49,和 LIGHT+ERW1041E = 3.28 ± 0.87,P < 0.01,n = 3),这两个组织与高血压的发生密切相关。我们的体外细胞试验表明,LIGHT 刺激触发了 AT1R 丰度的快速 TG2 依赖性增加(2 小时 LIGHT 处理后的相对 AT1R 水平:AT1R(WT)+TG2 = 2.21 ± 0.23,AT1R(Q315A)+TG2 = 0.18 ± 0.23,P < 0.05 与起始点 = 1,n = 2)和下游钙信号(NFAT 驱动的荧光素酶活性的倍数增加:盐水 = 0.02 ± 0.03,Ang II = 0.17 ± 0.08,LIGHT = 0.05 ± 0.04,LIGHT+Ang II = 0.90 ± 0.04(P < 0.01 与 Ang II),和 LIGHT+Ang II+ERW1041E = 0.15 ± 0.15(P < 0.01 与 LIGHT+Ang II),n = 3)。
我们的数据表明,TG2 在通过其稳定 AT1 受体和敏化 Ang II 的翻译后修饰将炎症与高血压联系起来方面发挥了重要的系统性作用。我们的发现还表明,TG2 抑制剂可作为一类新的心血管药物。