Computational Biology, GSK R&D, 1250 S. Collegeville Road, UP12-200, Collegeville, PA, USA.
Genetics, GSK R&D, 1250 S. Collegeville Road, UP12-200, Collegeville, PA, USA.
BMC Bioinformatics. 2019 Feb 8;20(1):69. doi: 10.1186/s12859-019-2664-1.
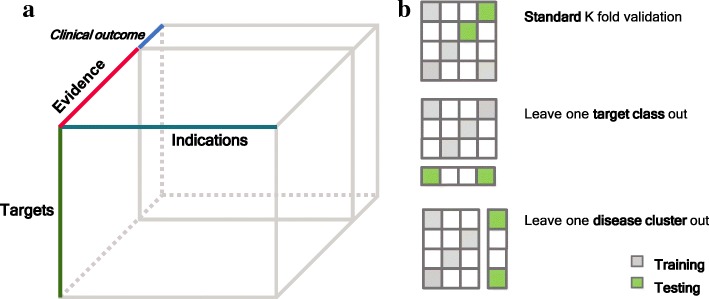
Determining which target to pursue is a challenging and error-prone first step in developing a therapeutic treatment for a disease, where missteps are potentially very costly given the long-time frames and high expenses of drug development. With current informatics technology and machine learning algorithms, it is now possible to computationally discover therapeutic hypotheses by predicting clinically promising drug targets based on the evidence associating drug targets with disease indications. We have collected this evidence from Open Targets and additional databases that covers 17 sources of evidence for target-indication association and represented the data as a tensor of 21,437 × 2211 × 17.
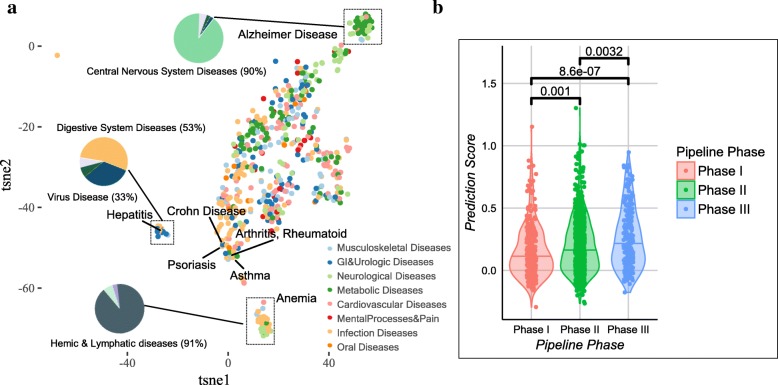
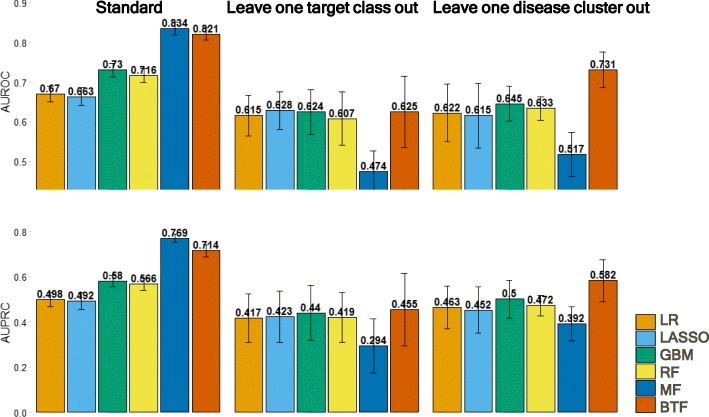
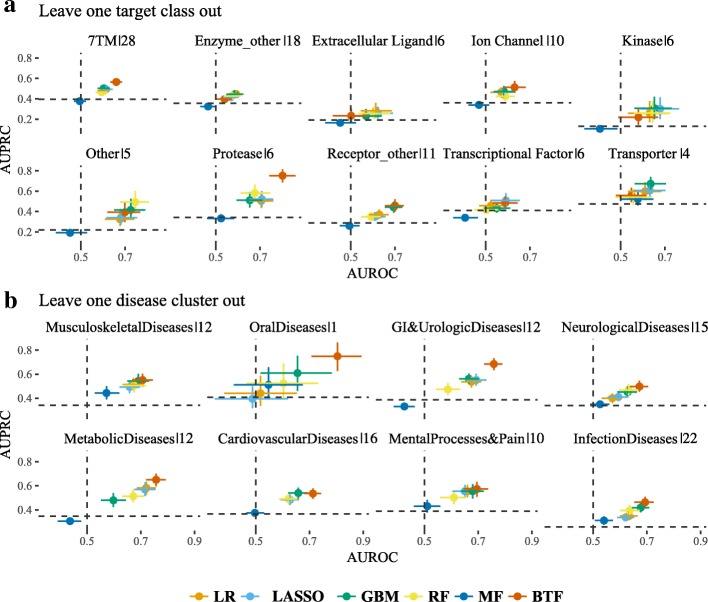
As a proof-of-concept, we identified examples of successes and failures of target-indication pairs in clinical trials across 875 targets and 574 disease indications to build a gold-standard data set of 6140 known clinical outcomes. We designed and executed three benchmarking strategies to examine the performance of multiple machine learning models: Logistic Regression, LASSO, Random Forest, Tensor Factorization and Gradient Boosting Machine. With 10-fold cross-validation, tensor factorization achieved AUROC = 0.82 ± 0.02 and AUPRC = 0.71 ± 0.03. Across multiple validation schemes, this was comparable or better than other methods.
In this work, we benchmarked a machine learning technique called tensor factorization for the problem of predicting clinical outcomes of therapeutic hypotheses. Results have shown that this method can achieve equal or better prediction performance compared with a variety of baseline models. We demonstrate one application of the method to predict outcomes of trials on novel indications of approved drug targets. This work can be expanded to targets and indications that have never been clinically tested and proposing novel target-indication hypotheses. Our proposed biologically-motivated cross-validation schemes provide insight into the robustness of the prediction performance. This has significant implications for all future methods that try to address this seminal problem in drug discovery.
在开发治疗疾病的治疗方法时,确定目标是一个具有挑战性且容易出错的第一步,因为药物开发的时间框架长且费用高,所以错误的决策代价可能非常高。借助当前的信息学技术和机器学习算法,现在可以通过预测基于将药物靶点与疾病适应症相关联的证据在临床上有前途的药物靶点,计算发现治疗假说。我们已经从 Open Targets 和其他涵盖 17 种药物靶点适应症关联证据来源的数据库中收集了这些证据,并将数据表示为 21437×2211×17 的张量。
作为概念验证,我们在 875 个靶点和 574 种疾病适应症的临床试验中确定了靶点-适应症对成功和失败的例子,以建立一个包含 6140 个已知临床结果的黄金标准数据集。我们设计并执行了三种基准测试策略来检查多种机器学习模型的性能:逻辑回归、LASSO、随机森林、张量分解和梯度提升机。通过 10 折交叉验证,张量分解的 AUROC=0.82±0.02,AUPRC=0.71±0.03。在多种验证方案中,这与其他方法相当或更好。
在这项工作中,我们对一种称为张量分解的机器学习技术进行了基准测试,以解决预测治疗假说临床结果的问题。结果表明,与各种基线模型相比,该方法可以实现相等或更好的预测性能。我们展示了该方法在预测已批准药物靶点新适应症试验结果中的一种应用。这项工作可以扩展到从未进行过临床测试的靶点和适应症,并提出新的靶点-适应症假说。我们提出的基于生物学的交叉验证方案为预测性能的稳健性提供了深入了解。这对所有试图解决药物发现中这一重要问题的未来方法都具有重要意义。