Cardiovascular Genetics, Department of Pediatrics, Centre Hospitalier Universitaire Sainte Justine Research Center, Université de Montréal, Montréal, Québec, Canada.
Department of Biochemistry and Molecular Medicine, Centre Hospitalier Universitaire Sainte Justine Research Center, Université de Montréal, Montréal, Québec, Canada.
Cell Mol Gastroenterol Hepatol. 2019;7(2):411-431. doi: 10.1016/j.jcmgh.2018.10.011. Epub 2018 Oct 24.
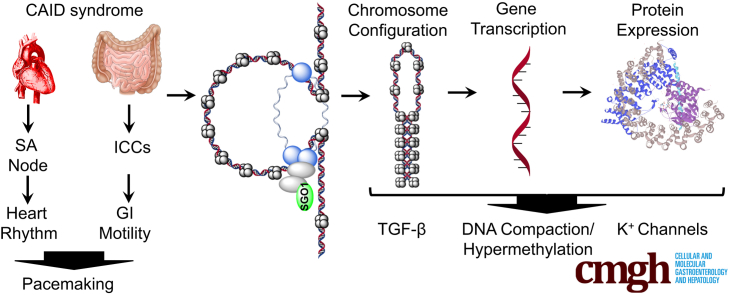
BACKGROUND & AIMS: A generalized human pacemaking syndrome, chronic atrial and intestinal dysrhythmia (CAID) (OMIM 616201), is caused by a homozygous SGO1 mutation (K23E), leading to chronic intestinal pseudo-obstruction and arrhythmias. Because CAID patients do not show phenotypes consistent with perturbation of known roles of SGO1, we hypothesized that noncanonical roles of SGO1 drive the clinical manifestations observed.
To identify a molecular signature for CAID syndrome, we achieved unbiased screens in cell lines and gut tissues from CAID patients vs wild-type controls. We performed RNA sequencing along with stable isotope labeling with amino acids in cell culture. In addition, we determined the genome-wide DNA methylation and chromatin accessibility signatures using reduced representative bisulfite sequencing and assay for transposase-accessible chromatin with high-throughput sequencing. Functional studies included patch-clamp, quantitation of transforming growth factor-β (TGF-β) signaling, and immunohistochemistry in CAID patient gut biopsy specimens.
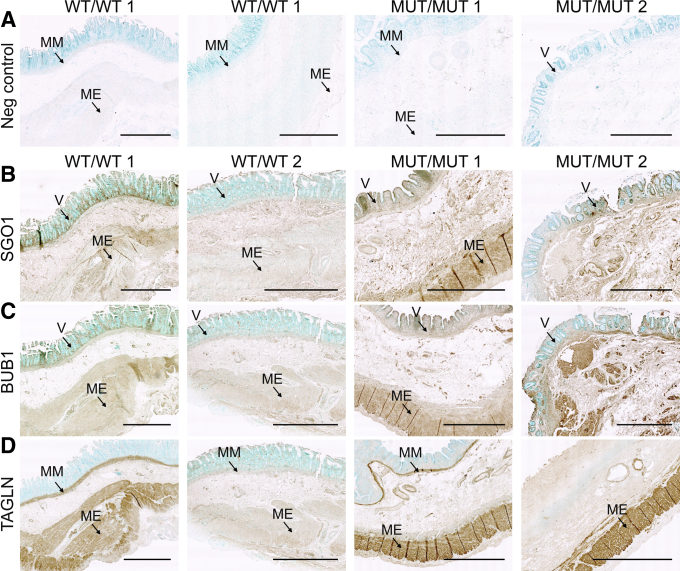
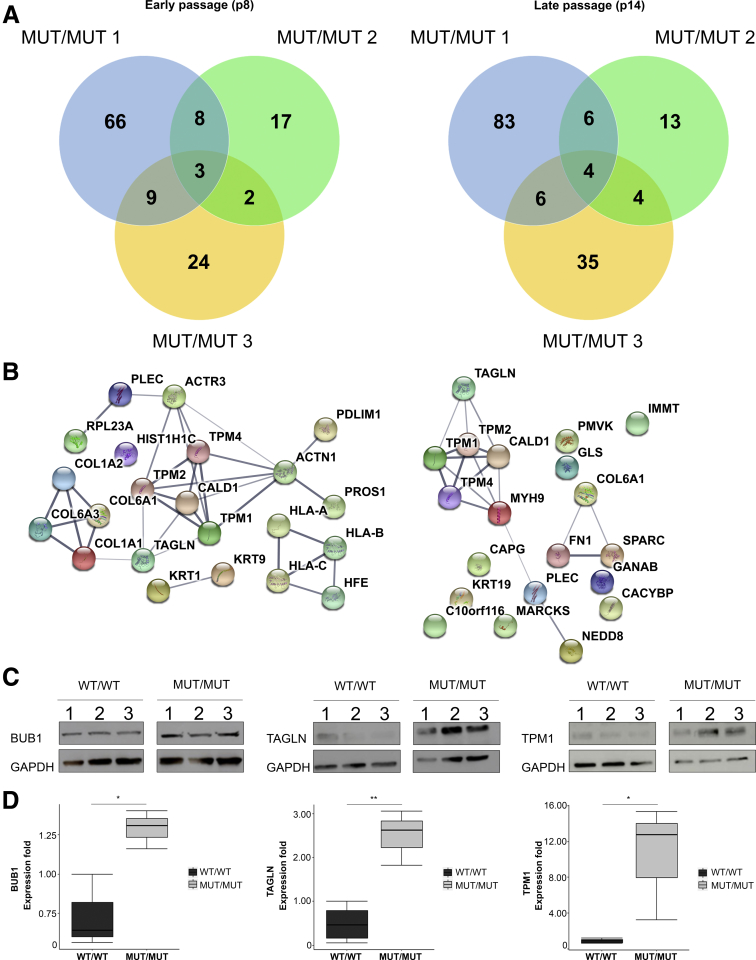
Proteome and transcriptome studies converge on cell-cycle regulation, cardiac conduction, and smooth muscle regulation as drivers of CAID syndrome. Specifically, the inward rectifier current, an important regulator of cellular function, was disrupted. Immunohistochemistry confirmed overexpression of Budding Uninhibited By Benzimidazoles 1 (BUB1) in patients, implicating the TGF-β pathway in CAID pathogenesis. Canonical TGF-β signaling was up-regulated and uncoupled from noncanonical signaling in CAID patients. Reduced representative bisulfite sequencing and assay for transposase-accessible chromatin with high-throughput sequencing experiments showed significant changes of chromatin states in CAID, pointing to epigenetic regulation as a possible pathologic mechanism.
Our findings point to impaired inward rectifier potassium current, dysregulation of canonical TGF-β signaling, and epigenetic regulation as potential drivers of intestinal and cardiac manifestations of CAID syndrome. Transcript profiling and genomics data are as follows: repository URL: https://www.ncbi.nlm.nih.gov/geo; SuperSeries GSE110612 was composed of the following subseries: GSE110309, GSE110576, and GSE110601.
一种广义的人类起搏综合征,即慢性心房和肠道节律紊乱(CAID)(OMIM 616201),是由 SGO1 基因(K23E)纯合突变引起的,导致慢性肠道假性肠梗阻和心律失常。由于 CAID 患者没有表现出与 SGO1 已知作用失调一致的表型,我们假设 SGO1 的非典型作用驱动了观察到的临床症状。
为了确定 CAID 综合征的分子特征,我们对 CAID 患者与野生型对照的细胞系和肠道组织进行了无偏筛选。我们在细胞培养中进行了 RNA 测序以及稳定同位素标记的氨基酸。此外,我们使用代表性降低的亚硫酸氢盐测序和高通量测序的转座酶可及染色质测定法,确定了全基因组的 DNA 甲基化和染色质可及性特征。功能研究包括 CAID 患者肠道活检标本的膜片钳、转化生长因子-β(TGF-β)信号的定量和免疫组织化学。
蛋白质组学和转录组学研究集中在细胞周期调控、心脏传导和平滑肌调控上,这些都是 CAID 综合征的驱动因素。具体来说,一种重要的细胞功能调节的内向整流电流受到了破坏。免疫组织化学证实患者中 Budding Uninhibited By Benzimidazoles 1(BUB1)的过度表达,提示 TGF-β 通路参与了 CAID 的发病机制。CAID 患者中经典 TGF-β 信号被上调并与非经典信号解偶联。代表性降低的亚硫酸氢盐测序和高通量测序的转座酶可及染色质测定实验显示 CAID 中染色质状态有显著变化,提示表观遗传调控可能是一种潜在的病理机制。
我们的发现表明,内向整流钾电流受损、经典 TGF-β 信号失调以及表观遗传调控可能是 CAID 综合征肠道和心脏表现的潜在驱动因素。转录谱和基因组学数据如下:存储库 URL:https://www.ncbi.nlm.nih.gov/geo;SuperSeries GSE110612 由以下子系列组成:GSE110309、GSE110576 和 GSE110601。