National Institutes of Health Undiagnosed Diseases Program, Common Fund, Office of the Director, Bethesda, Maryland.
Office of the Clinical Director, National Human Genome Research Institute National Human Genome Research Institute, National Institutes of Health, Bethesda, Maryland.
Hum Mutat. 2019 May;40(5):532-538. doi: 10.1002/humu.23722. Epub 2019 Mar 12.
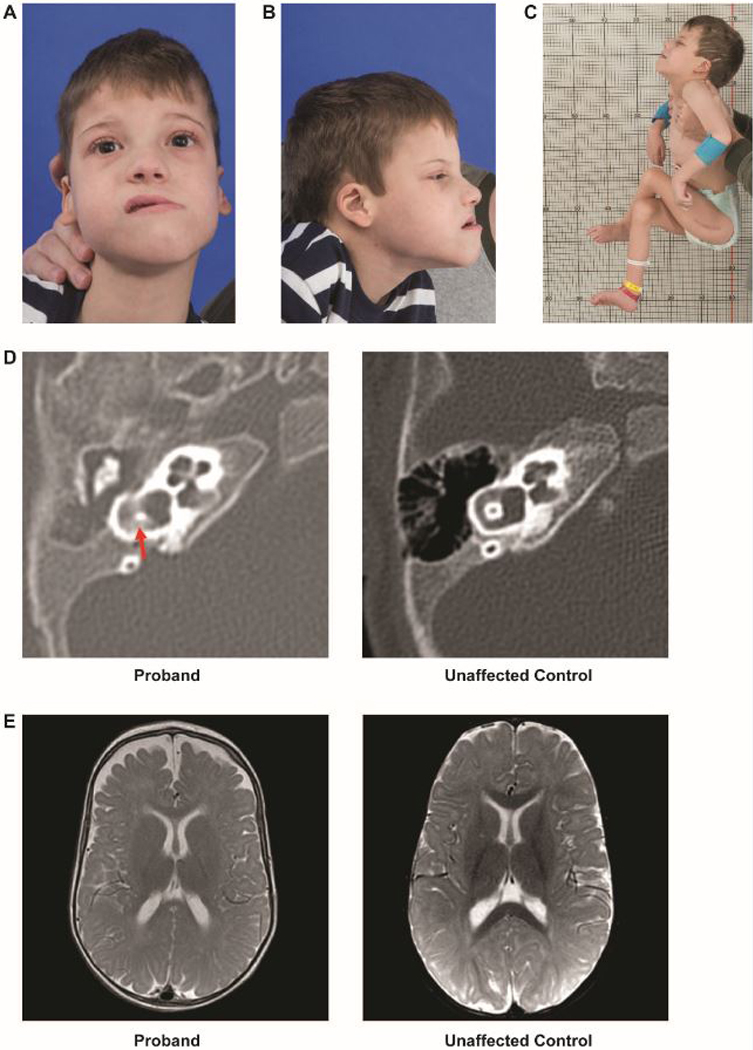
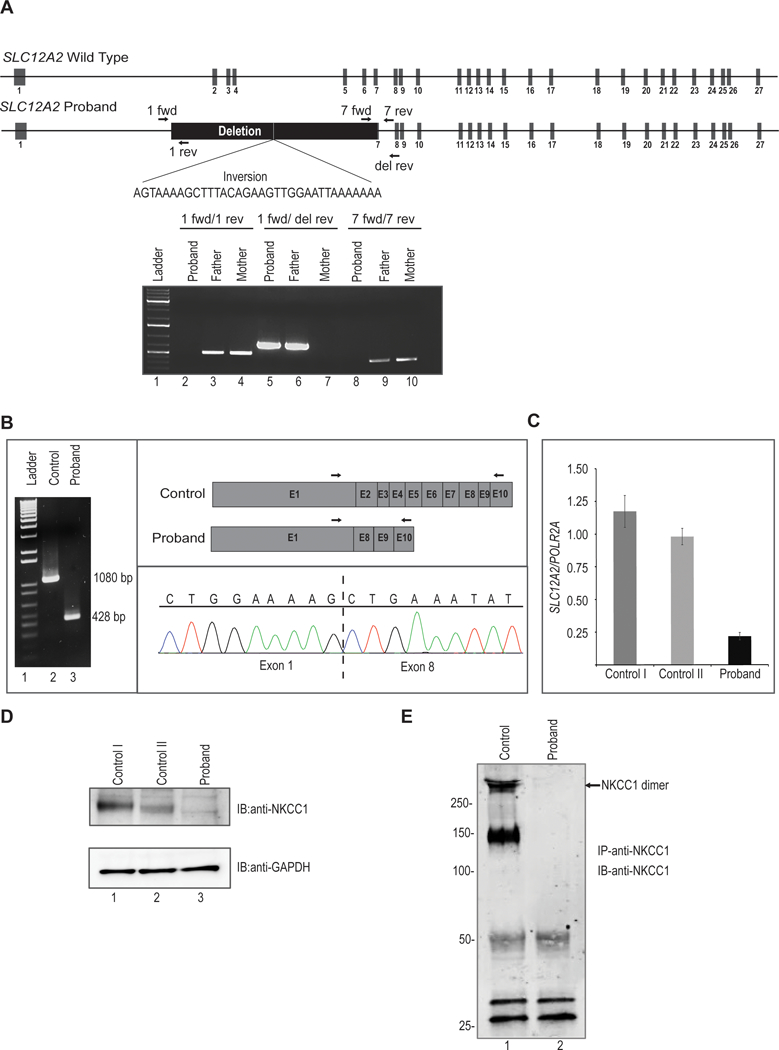
Syndromic sensorineural hearing loss is multigenic and associated with malformations of the ear and other organ systems. Herein we describe a child admitted to the NIH Undiagnosed Diseases Program with global developmental delay, sensorineural hearing loss, gastrointestinal abnormalities, and absent salivation. Next-generation sequencing revealed a uniparental isodisomy in chromosome 5, and a 22 kb homozygous deletion in SLC12A2, which encodes for sodium, potassium, and chloride transporter in the basolateral membrane of secretory epithelia. Functional studies using patient-derived fibroblasts showed truncated SLC12A2 transcripts and markedly reduced protein abundance when compared with control. Loss of Slc12a2 in mice has been shown to lead to deafness, abnormal neuronal growth and migration, severe gastrointestinal abnormalities, and absent salivation. Together with the described phenotype of the Slc12a2-knockout mouse model, our results suggest that the absence of functional SLC12A2 causes a new genetic syndrome and is crucial for the development of auditory, neurologic, and gastrointestinal tissues.
综合征性感觉神经性听力损失是多基因的,并与耳朵和其他器官系统的畸形有关。在此,我们描述了一名因全身发育迟缓、感觉神经性听力损失、胃肠道异常和无唾液分泌而被纳入 NIH 未确诊疾病计划的儿童。下一代测序显示 5 号染色体单亲二体性和 SLC12A2 中的 22kb 纯合缺失,该缺失编码分泌上皮基底外侧膜中的钠、钾和氯转运体。使用患者来源的成纤维细胞进行的功能研究表明,与对照相比,SLC12A2 的截断转录本和明显减少的蛋白丰度。已经表明,在小鼠中缺失 Slc12a2 会导致耳聋、异常神经元生长和迁移、严重的胃肠道异常和无唾液分泌。结合 Slc12a2 基因敲除小鼠模型的描述表型,我们的结果表明,功能性 SLC12A2 的缺失导致了一种新的遗传综合征,并对听觉、神经和胃肠道组织的发育至关重要。