Computation-based Science and Technology Research Center CaSToRC , The Cyprus Institute , 20 Konstaninou Kavafi Street , 2121 Aglantzia, Nicosia , Cyprus.
Departments of Physics , Faculty of Mathematics, Computer Science and Natural Sciences, Aachen University , Otto-Blumenthal Straße , 52062 Aachen , Germany.
J Chem Theory Comput. 2019 Mar 12;15(3):2101-2109. doi: 10.1021/acs.jctc.9b00040. Epub 2019 Feb 28.
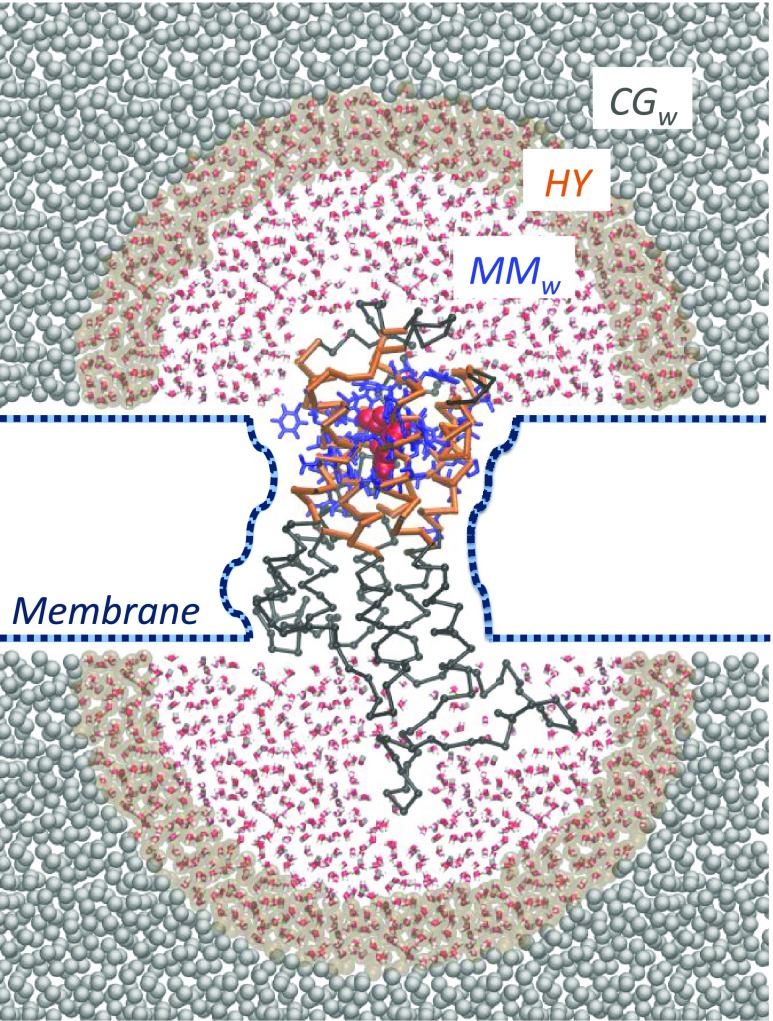
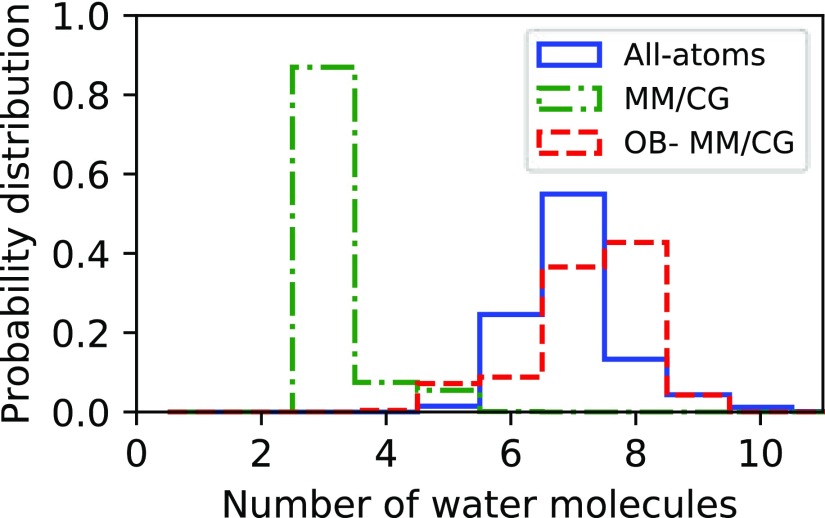
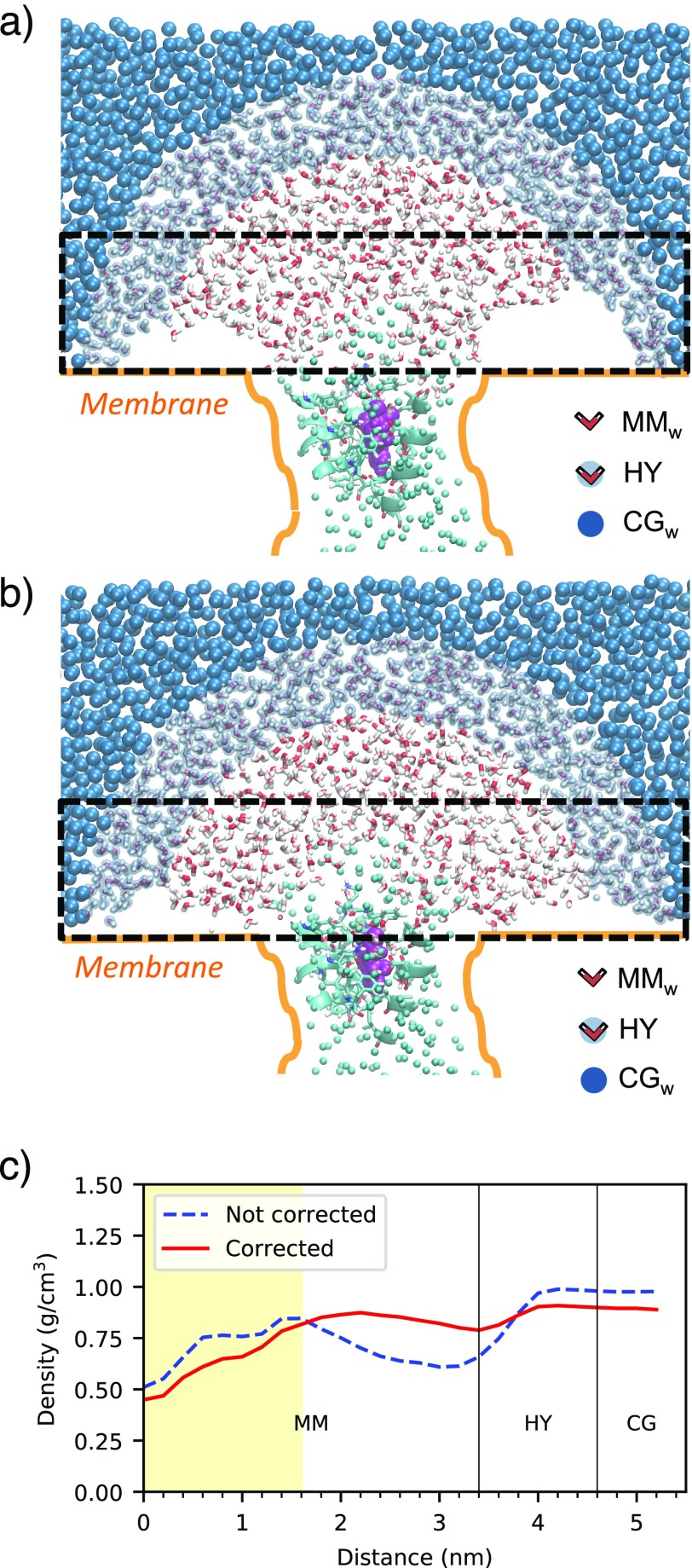
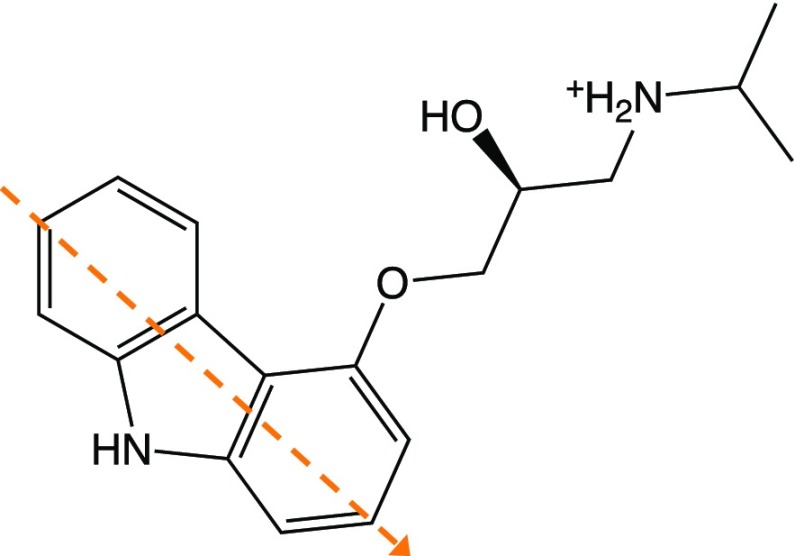
G-protein-coupled receptors (GPCRs) constitute as much as 30% of the overall proteins targeted by FDA-approved drugs. However, paucity of structural experimental information and low sequence identity between members of the family impair the reliability of traditional docking approaches and atomistic molecular dynamics simulations for in silico pharmacological applications. We present here a dual-resolution approach tailored for such low-resolution models. It couples a hybrid molecular mechanics/coarse-grained (MM/CG) scheme, previously developed by us for GPCR-ligand complexes, with a Hamiltonian-based adaptive resolution scheme (H-AdResS) for the solvent. This dual-resolution approach removes potentially inaccurate atomistic details from the model while building a rigorous statistical ensemble-the grand canonical one-in the high-resolution region. We validate the method on a well-studied GPCR-ligand complex, for which the 3D structure is known, against atomistic simulations. This implementation paves the way for future accurate in silico studies of low-resolution ligand/GPCRs models.
G 蛋白偶联受体(GPCRs)构成了经美国食品和药物管理局批准的药物所针对的全部蛋白质的 30%左右。然而,结构实验信息的缺乏以及家族成员之间的低序列同一性,降低了传统对接方法和原子分子动力学模拟在计算机药理学应用中的可靠性。我们在这里提出了一种针对这种低分辨率模型的双重分辨率方法。它将我们之前为 GPCR-配体复合物开发的混合力学/粗粒化(MM/CG)方案与基于哈密顿量的自适应分辨率方案(H-AdResS)相结合,用于溶剂。这种双重分辨率方法在构建高分辨率区域中的严格统计系综——正则系综时,从模型中去除潜在的不准确原子细节。我们使用已知 3D 结构的经过充分研究的 GPCR-配体复合物对该方法进行了验证,结果与原子模拟一致。这种实现为未来对低分辨率配体/GPCR 模型进行准确的计算机研究铺平了道路。