Department of Biology, Massachusetts Institute of Technology, Cambridge, MA 02139, USA.
Department of Computer Science, Dartmouth College, Hanover, NH 03755, USA.
Structure. 2019 Apr 2;27(4):606-617.e5. doi: 10.1016/j.str.2019.01.008. Epub 2019 Feb 14.
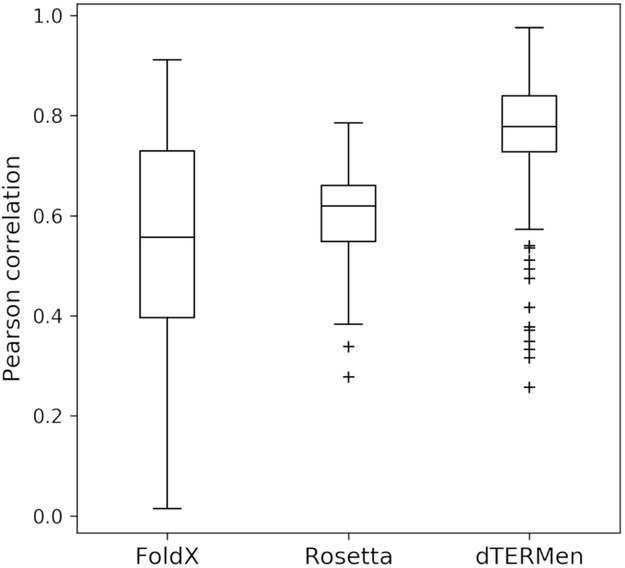
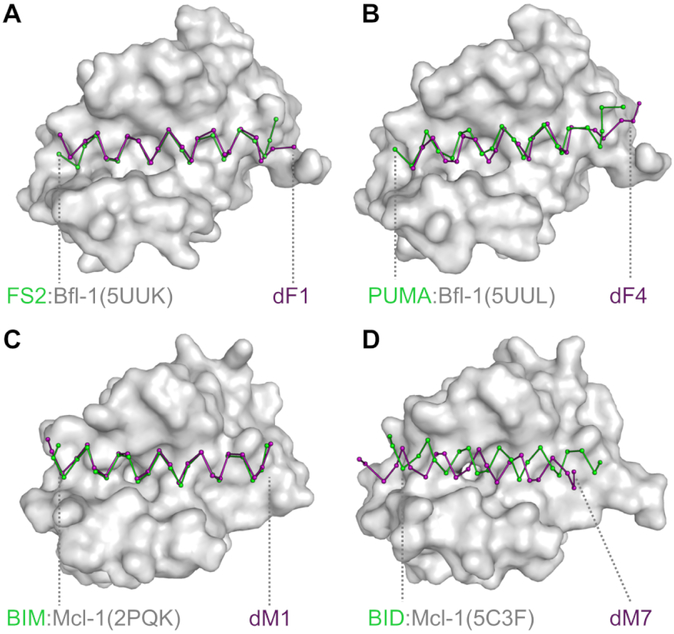
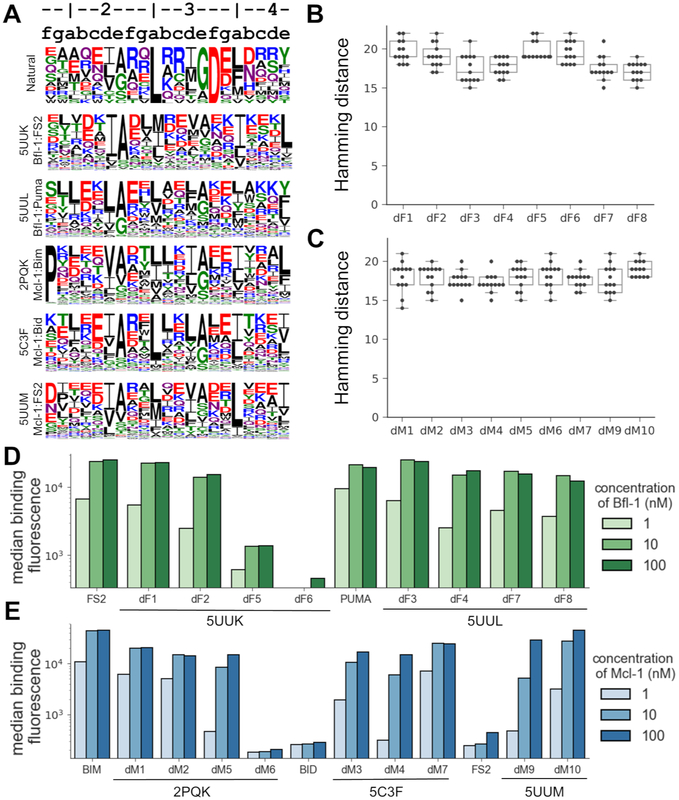
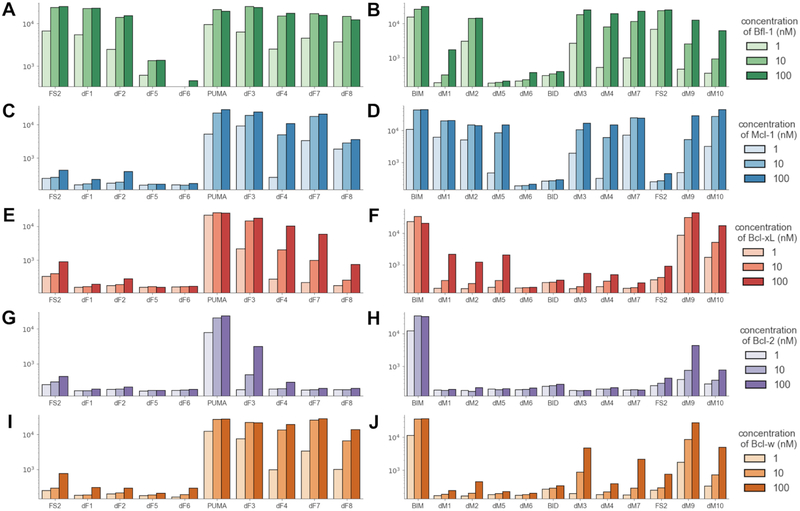
Understanding the relationship between protein sequence and structure well enough to design new proteins with desired functions is a longstanding goal in protein science. Here, we show that recurring tertiary structural motifs (TERMs) in the PDB provide rich information for protein-peptide interaction prediction and design. TERM statistics can be used to predict peptide binding energies for Bcl-2 family proteins as accurately as widely used structure-based tools. Furthermore, design using TERM energies (dTERMen) rapidly and reliably generates high-affinity peptide binders of anti-apoptotic proteins Bfl-1 and Mcl-1 with just 15%-38% sequence identity to any known native Bcl-2 family protein ligand. High-resolution structures of four designed peptides bound to their targets provide opportunities to analyze the strengths and limitations of the computational design method. Our results support dTERMen as a powerful approach that can complement existing tools for protein engineering.
充分了解蛋白质序列和结构之间的关系,以便设计具有预期功能的新蛋白质,这是蛋白质科学的一个长期目标。在这里,我们表明 PDB 中的重复三级结构基序 (TERM) 为蛋白质-肽相互作用的预测和设计提供了丰富的信息。TERM 统计信息可用于准确预测 Bcl-2 家族蛋白的肽结合能,与广泛使用的基于结构的工具一样准确。此外,使用 TERM 能量进行设计 (dTERMen) 可以快速可靠地生成高亲和力的抗凋亡蛋白 Bfl-1 和 Mcl-1 的肽结合物,与任何已知的天然 Bcl-2 家族蛋白配体的序列同一性仅为 15%-38%。四个设计肽与它们的靶标的高分辨率结构提供了分析计算设计方法的优缺点的机会。我们的结果支持 dTERMen 作为一种强大的方法,可以补充现有蛋白质工程工具。