Hamlin Trevor A, Levandowski Brian J, Narsaria Ayush K, Houk Kendall N, Bickelhaupt F Matthias
Department of Theoretical Chemistry, Amsterdam Center for Multiscale Modeling (ACMM), Vrije Universiteit Amsterdam, De Boelelaan 1083, 1081 HV, Amsterdam, The Netherlands.
Department of Chemistry and Biochemistry, University of California, Los Angeles, CA, 90095, USA.
Chemistry. 2019 May 2;25(25):6342-6348. doi: 10.1002/chem.201900295. Epub 2019 Mar 27.
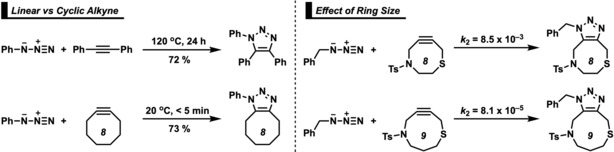
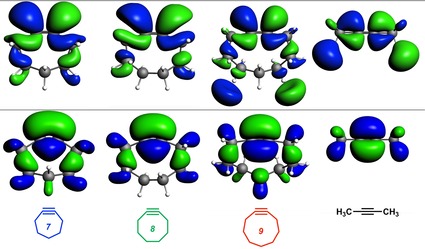
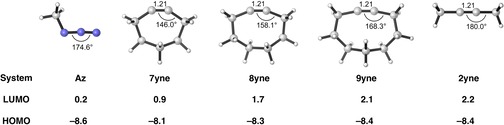
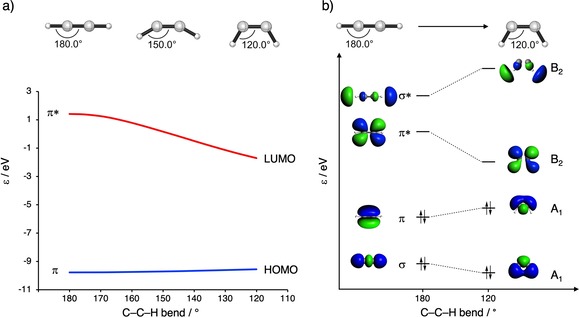
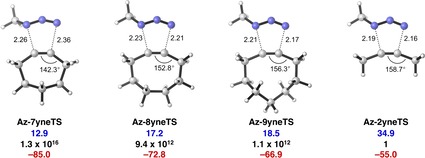
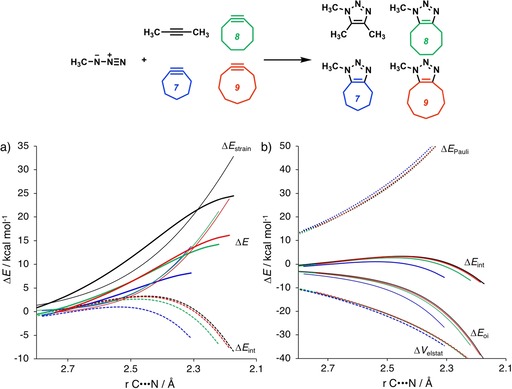
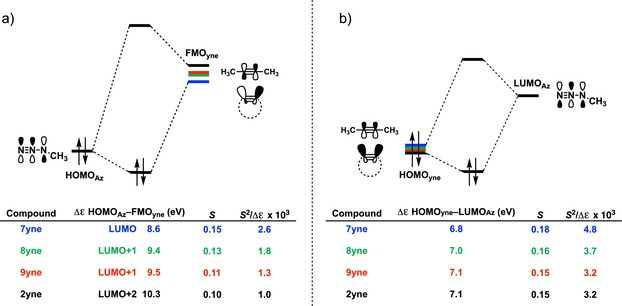
The reactivities of 2-butyne, cycloheptyne, cyclooctyne, and cyclononyne in the 1,3-dipolar cycloaddition reaction with methyl azide were evaluated through DFT calculations at the M06-2X/6-311++G(d)//M06-2X/6-31+G(d) level of theory. Computed activation free energies for the cycloadditions of cycloalkynes are 16.5-22.0 kcal mol lower in energy than that of the acyclic 2-butyne. The strained or predistorted nature of cycloalkynes is often solely used to rationalize this significant rate enhancement. Our distortion/interaction-activation strain analysis has been revealed that the degree of geometrical predistortion of the cycloalkyne ground-state geometries acts to enhance reactivity compared with that of acyclic alkynes through three distinct mechanisms, not only due to (i) a reduced strain or distortion energy, but also to (ii) a smaller HOMO-LUMO gap, and (iii) an enhanced orbital overlap, which both contribute to more stabilizing orbital interactions.
通过在M06 - 2X/6 - 311++G(d)//M06 - 2X/6 - 31+G(d)理论水平下的密度泛函理论(DFT)计算,评估了2 - 丁炔、环庚炔、环辛炔和环壬炔与叠氮甲烷在1,3 - 偶极环加成反应中的反应活性。计算得出,环炔烃环加成反应的活化自由能比无环的2 - 丁炔低16.5 - 22.0千卡/摩尔。环炔烃的应变或预扭曲性质通常仅用于解释这种显著的速率增强。我们的扭曲/相互作用 - 活化应变分析表明,与无环炔烃相比,环炔烃基态几何结构的几何预扭曲程度通过三种不同机制提高了反应活性,这不仅是由于(i)应变或扭曲能降低,还由于(ii)较小的最高已占分子轨道(HOMO) - 最低未占分子轨道(LUMO)能隙,以及(iii)增强的轨道重叠,这两者都有助于形成更稳定的轨道相互作用。