Department of Physics and Astronomy, University of Texas Rio Grande Valley, Edinburg, TX 78539, USA.
Achieve Early College High School, McAllen, TX 78501, USA.
Molecules. 2019 Feb 19;24(4):754. doi: 10.3390/molecules24040754.
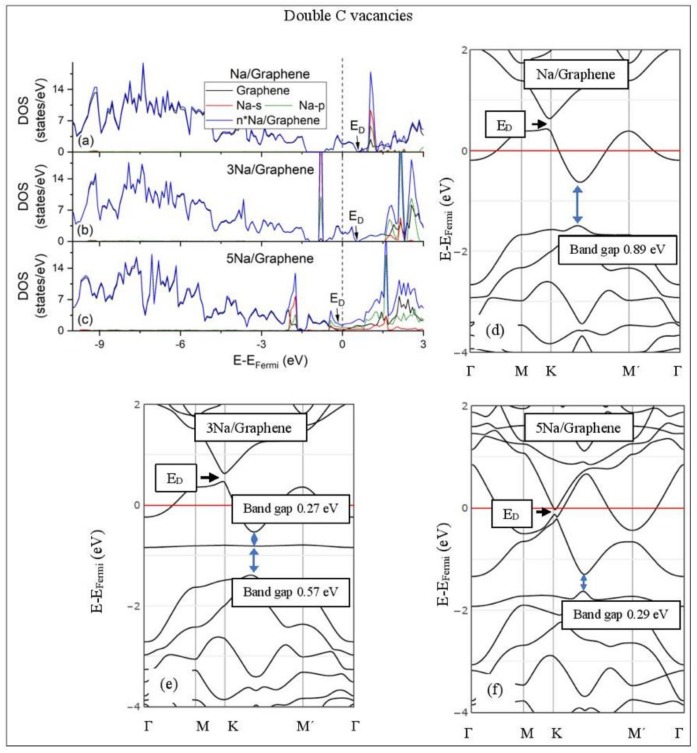
Adsorption of Li and Na on pristine and defective graphene and graphene oxide (GO) is studied using density functional theory (DFT) structural and electronic calculations, quantum theory of atoms in molecules (QTAIM), and electron localization function (ELF) analyses. DFT calculations show that Li and Na adsorptions on pristine graphene are not stable at all metal coverages examined here. However, the presence of defects on graphene support stabilizes both Li and Na adsorptions. Increased Li and Na coverages cause metal nucleation and weaken adsorption. Defective graphene is associated with the presence of band gaps and, thus, Li and Na adsorptions can be used to tune these gaps. Electronic calculations show that Li⁻ and Na⁻graphene interactions are Coulombic: as Li and Na coverages increase, the metal valences partially hybridize with the graphene bands and weaken metal⁻graphene support interactions. However, for Li adsorption on single vacancy graphene, QTAIM, ELF, and overlap populations calculations show that the Li-C bond has some covalent character. The Li and Na adsorptions on GO are significantly stronger than on graphene and strengthen upon increased coverages. This is due to Li and Na forming bonds with both carbon and oxygen GO atoms. QTAIM and ELF are used to analyze the metal⁻C and metal⁻metal bonds (when metal nucleation is present). The Li and Na clusters may contain both covalent and metallic intra metal⁻metal bonds: This effect is related to the adsorption support selection. ELF bifurcation diagrams show individual metal⁻C and metal⁻metal interactions, as Li and Na are adsorbed on graphene and GO, at the metal coverages examined here.
使用密度泛函理论(DFT)结构和电子计算、原子在分子中的量子理论(QTAIM)和电子定域函数(ELF)分析研究了原始和有缺陷的石墨烯和氧化石墨烯(GO)上 Li 和 Na 的吸附。DFT 计算表明,在此处研究的所有金属覆盖率下,Li 和 Na 在原始石墨烯上的吸附都不稳定。然而,石墨烯上缺陷的存在稳定了 Li 和 Na 的吸附。Li 和 Na 的覆盖增加会导致金属成核并削弱吸附。有缺陷的石墨烯与带隙的存在有关,因此,Li 和 Na 的吸附可用于调节这些带隙。电子计算表明,Li⁻和 Na⁻与石墨烯的相互作用是库仑的:随着 Li 和 Na 覆盖率的增加,金属价态部分与石墨烯带杂化并削弱金属⁻石墨烯支撑相互作用。然而,对于单空位石墨烯上的 Li 吸附,QTAIM、ELF 和重叠人口计算表明,Li-C 键具有一定的共价特征。Li 和 Na 在 GO 上的吸附明显强于在石墨烯上,并且随着覆盖率的增加而增强。这是由于 Li 和 Na 与 GO 的碳原子和氧原子形成键。QTAIM 和 ELF 用于分析金属⁻C 和金属⁻金属键(当存在金属成核时)。Li 和 Na 簇可能包含共价和金属内金属⁻金属键:这种效应与吸附支撑的选择有关。ELF 分叉图显示了单个金属⁻C 和金属⁻金属相互作用,当 Li 和 Na 吸附在石墨烯和 GO 上时,在此处研究的金属覆盖率下。