Centre for Rare Diseases, Aarhus University Hospital, Aarhus, Denmark.
Royal Manchester Children's Hospital, Manchester, UK.
Orphanet J Rare Dis. 2019 Feb 26;14(1):58. doi: 10.1186/s13023-019-1014-8.
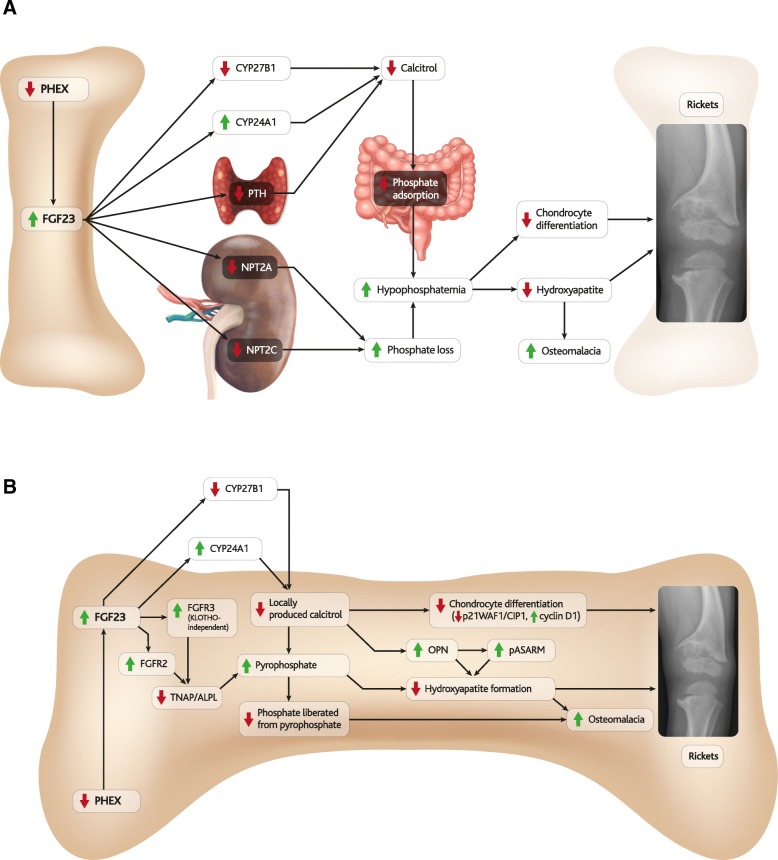
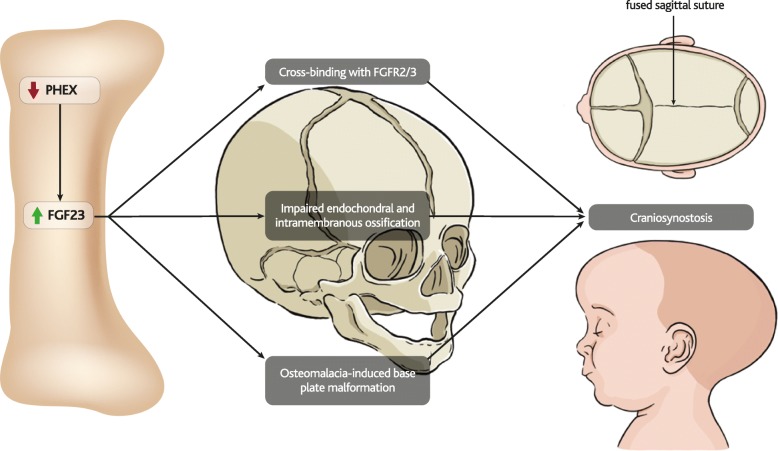
X-linked hypophosphatemia (XLH) is an inherited disease of phosphate metabolism in which inactivating mutations of the Phosphate Regulating Endopeptidase Homolog, X-Linked (PHEX) gene lead to local and systemic effects including impaired growth, rickets, osteomalacia, bone abnormalities, bone pain, spontaneous dental abscesses, hearing difficulties, enthesopathy, osteoarthritis, and muscular dysfunction. Patients with XLH present with elevated levels of fibroblast growth factor 23 (FGF23), which is thought to mediate many of the aforementioned manifestations of the disease. Elevated FGF23 has also been observed in many other diseases of hypophosphatemia, and a range of animal models have been developed to study these diseases, yet the role of FGF23 in the pathophysiology of XLH is incompletely understood.
The role of FGF23 in the pathophysiology of XLH is here reviewed by describing what is known about phenotypes associated with various PHEX mutations, animal models of XLH, and non-nutritional diseases of hypophosphatemia, and by presenting molecular pathways that have been proposed to contribute to manifestations of XLH.
The pathophysiology of XLH is complex, involving a range of molecular pathways that variously contribute to different manifestations of the disease. Hypophosphatemia due to elevated FGF23 is the most obvious contributor, however localised fluctuations in tissue non-specific alkaline phosphatase (TNAP), pyrophosphate, calcitriol and direct effects of FGF23 have been observed to be associated with certain manifestations.
By describing what is known about these pathways, this review highlights key areas for future research that would contribute to the understanding and clinical treatment of non-nutritional diseases of hypophosphatemia, particularly XLH.
X 连锁低磷血症(XLH)是一种遗传性磷代谢疾病,其致病基因 Phosphate Regulating Endopeptidase Homolog, X-Linked(PHEX)发生失活突变,导致包括生长障碍、佝偻病、骨软化症、骨骼异常、骨痛、自发性齿龈脓肿、听力障碍、肌腱病、骨关节炎和肌肉功能障碍在内的局部和全身效应。XLH 患者的成纤维细胞生长因子 23(FGF23)水平升高,这被认为介导了疾病的许多上述表现。在许多其他低磷血症疾病中也观察到了升高的 FGF23,并且已经开发了一系列动物模型来研究这些疾病,但 FGF23 在 XLH 病理生理学中的作用尚未完全阐明。
通过描述与各种 PHEX 突变相关的表型、XLH 动物模型以及非营养性低磷血症疾病,以及提出有助于 XLH 表现的分子途径,来综述 FGF23 在 XLH 病理生理学中的作用。
XLH 的病理生理学很复杂,涉及一系列分子途径,这些途径不同程度地导致疾病的不同表现。由于 FGF23 升高导致的低磷血症是最明显的原因,但局部组织非特异性碱性磷酸酶(TNAP)、焦磷酸盐、骨化三醇和 FGF23 的直接作用波动与某些表现有关。
通过描述这些途径的已知内容,本综述突出了未来研究的关键领域,这将有助于理解和临床治疗非营养性低磷血症,特别是 XLH。