Xia Hong, Huang Xiangjun, Xu Hongbo, Zhou Yong-An, Gong Lina, Yang Zhijian, Lv Jingyan, Deng Hao
Third Xiangya Hospital, Center for Experimental Medicine and Department of Neurology, Central South University, Changsha, Hunan, China.
First Affiliated Hospital, Department of General Surgery, Hunan University, Changsha, Hunan, China.
Genet Mol Biol. 2019 Jan-Mar;42(1):48-51. doi: 10.1590/1678-4685-gmb-2017-0318. Epub 2019 Feb 28.
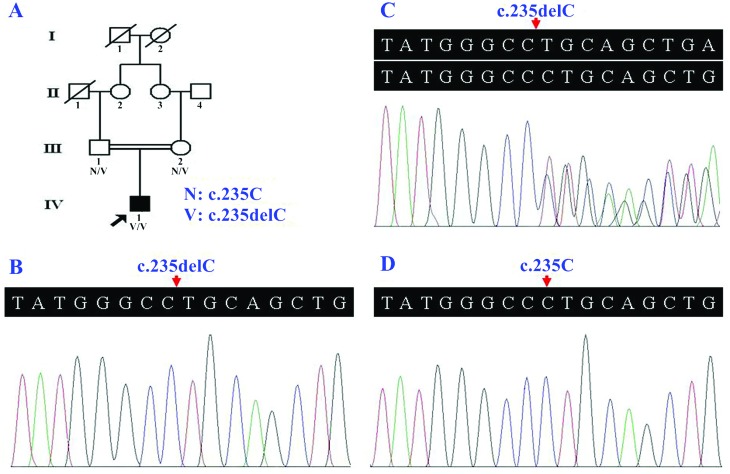
Autosomal recessive nonsyndromic hearing loss (ARNSHL) is a genetically heterogeneous neurosensory disorder, usually characterized by congenital or prelingual hearing loss. We report a Han Chinese male, born to consanguineous parents, presenting with nonsyndromic sensorineural hearing loss, whose clinical phenotype was also consistent with auditory neuropathy spectrum disorder (ANSD). After exome sequencing, a gap junction protein beta 2 gene (GJB2) c.235delC variant in the homozygous state was detected in the patient. Both parents were heterozygous for this variant, as documented by Sanger sequencing. The known pathogenic GJB2 c.235delC variant was not detected in 200 healthy controls. It is predicted to be a disease-causing alteration by generating a truncated protein p.(L79Cfs*3), disturbing the appropriate folding and/or oligomerization of connexins and leading to defective gap junction channels. This study shows that the association of homozygosity of the GJB2 c.235delC variant with ARNSHL and ANSD in a patient.
常染色体隐性非综合征性听力损失(ARNSHL)是一种具有遗传异质性的神经感觉障碍,通常表现为先天性或语前听力损失。我们报告了一名汉族男性,其父母为近亲结婚,患有非综合征性感音神经性听力损失,其临床表型也与听觉神经病谱系障碍(ANSD)一致。经外显子组测序,在该患者中检测到纯合状态的缝隙连接蛋白β2基因(GJB2)c.235delC变异。经桑格测序证实,其父母均为该变异的杂合子。在200名健康对照中未检测到已知的致病性GJB2 c.235delC变异。预计该变异会通过产生截短蛋白p.(L79Cfs*3)导致致病改变,干扰连接蛋白的正确折叠和/或寡聚化,并导致缝隙连接通道缺陷。本研究显示了一名患者中GJB2 c.235delC变异纯合性与ARNSHL和ANSD的关联。