Max Planck Institute for Informatics, Saarland Informatics Campus, 66123, Saarbrücken, Germany.
Present Address: Department of Genetics, Stanford University School of Medicine, Stanford, CA, 94305, USA.
Genome Biol. 2019 Mar 14;20(1):55. doi: 10.1186/s13059-019-1664-9.
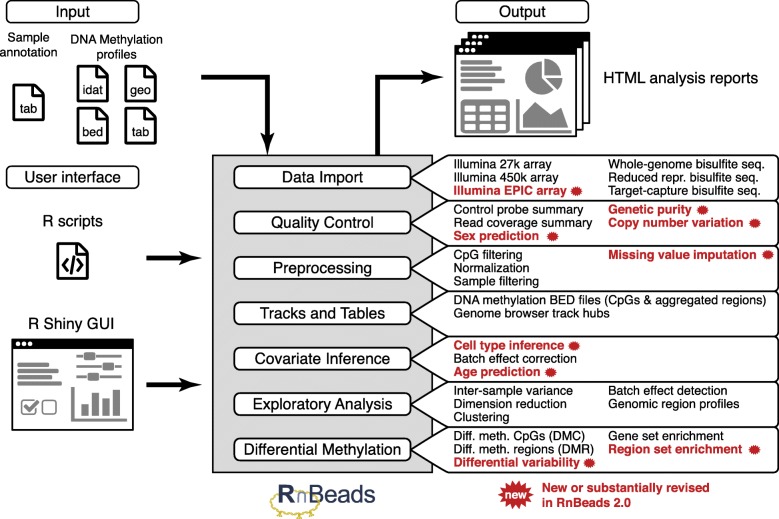
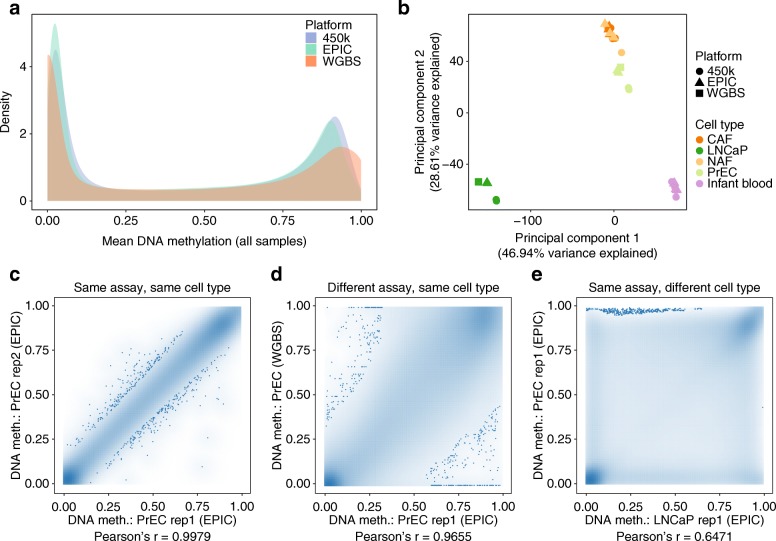
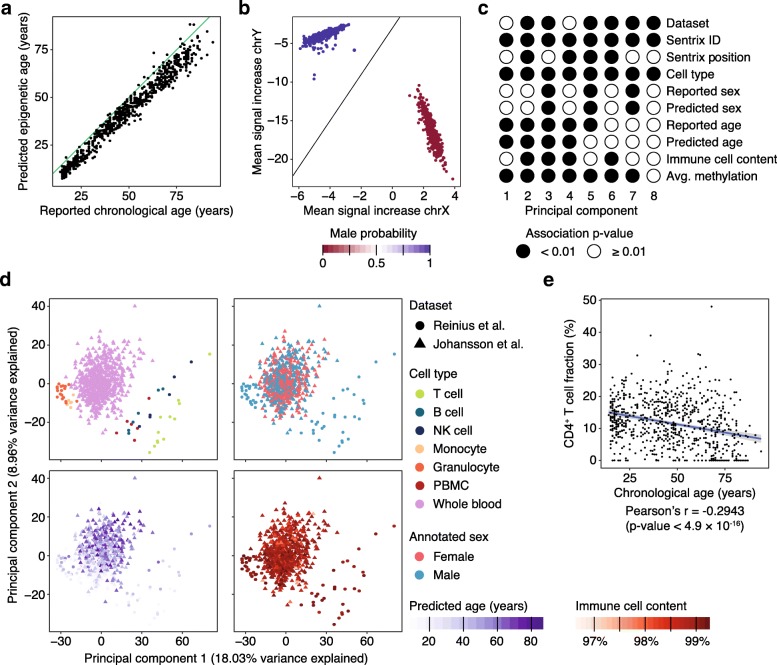
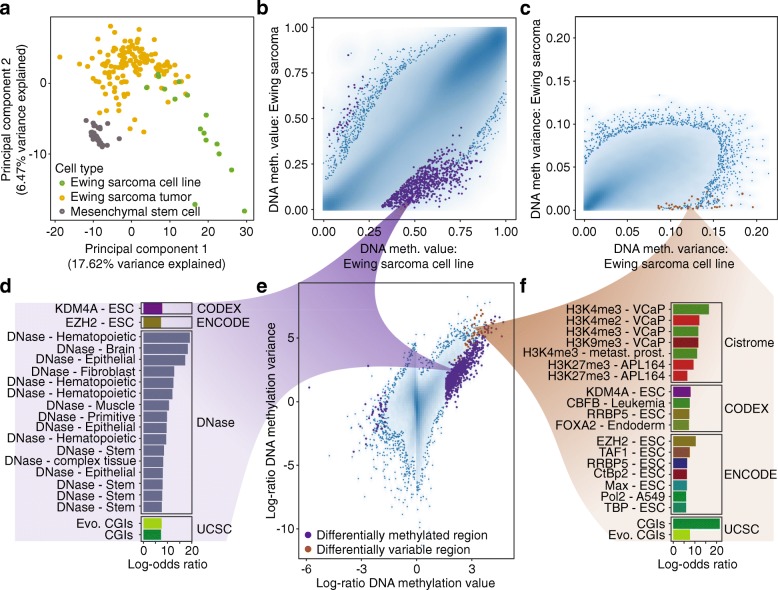
DNA methylation is a widely investigated epigenetic mark with important roles in development and disease. High-throughput assays enable genome-scale DNA methylation analysis in large numbers of samples. Here, we describe a new version of our RnBeads software - an R/Bioconductor package that implements start-to-finish analysis workflows for Infinium microarrays and various types of bisulfite sequencing. RnBeads 2.0 ( https://rnbeads.org/ ) provides additional data types and analysis methods, new functionality for interpreting DNA methylation differences, improved usability with a novel graphical user interface, and better use of computational resources. We demonstrate RnBeads 2.0 in four re-runnable use cases focusing on cell differentiation and cancer.
DNA 甲基化是一种广泛研究的表观遗传标记,在发育和疾病中具有重要作用。高通量检测方法可实现大量样本的全基因组 DNA 甲基化分析。在这里,我们描述了我们的 RnBeads 软件的新版本 - 一个 R/Bioconductor 包,它实现了针对 Infinium 微阵列和各种类型的亚硫酸氢盐测序的端到端分析工作流程。RnBeads 2.0(https://rnbeads.org/)提供了额外的数据类型和分析方法、用于解释 DNA 甲基化差异的新功能、具有新颖图形用户界面的改进可用性以及更好地利用计算资源。我们在四个可重复使用的用例中展示了 RnBeads 2.0,这些用例重点关注细胞分化和癌症。