Institute for Integrative Biology of the Cell (I2BC), CEA, CNRS, Université Paris-Sud, Université Paris-Saclay, Gif-Sur-Yvette, France.
BMC Biotechnol. 2019 Mar 20;19(1):18. doi: 10.1186/s12896-019-0509-7.
The CRISPR/Cas (clustered regularly interspaced short palindromic repeat and CRISPR-associated nucleases) based technologies have revolutionized genome engineering. While their use for prokaryotic genome editing is expanding, some limitations remain such as possible off-target effects and design constraints. These are compounded when performing systematic genome editing at distinct loci or when targeting repeated sequences (e.g. multicopy genes or mobile genetic elements). To overcome these limitations, we designed an approach using the same sgRNA and CRISPR-Cas9 system to independently perform gene editing at different loci.
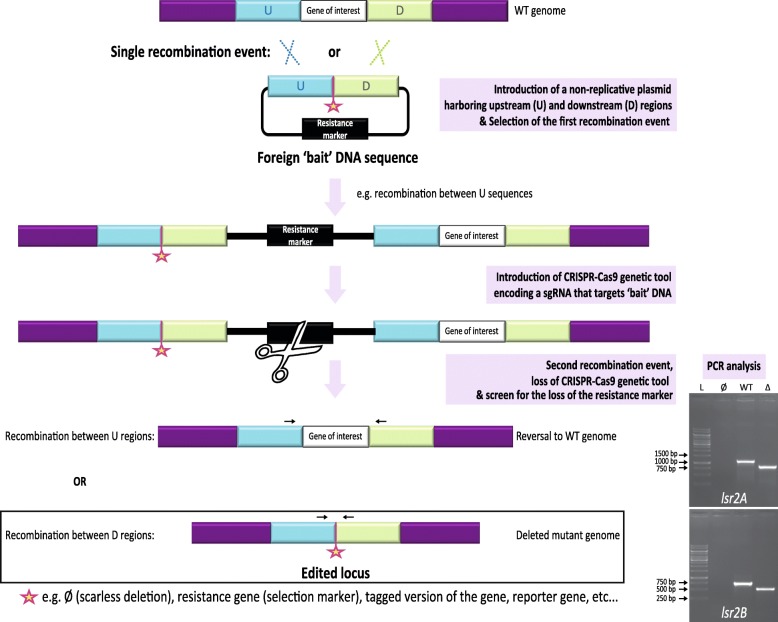
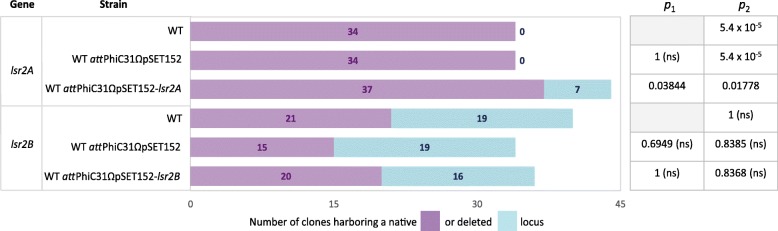
We developed a two-step procedure based on the introduction by homologous recombination of 'bait' DNA at the vicinity of a gene copy of interest before inducing CRISPR-Cas9 activity. The introduction of a genetic tool encoding a CRISPR-Cas9 complex targeting this 'bait' DNA induces a double strand break near the copy of interest. Its repair by homologous recombination can lead either to reversion or gene copy-specific editing. The relative frequencies of these events are linked to the impact of gene editing on cell fitness. In our study, we used this technology to successfully delete the native copies of two xenogeneic silencers lsr2 paralogs in Streptomyces ambofaciens. We observed that one of these paralogs is a candidate-essential gene since its native locus can be deleted only in the presence of an extra copy.
By targeting 'bait' DNA, we designed a 'generic' CRISPR-Cas9 toolkit that can be used to edit different loci. The differential action of this CRISPR-Cas9 system is exclusively based on the specific recombination between regions surrounding the gene copy of interest. This approach is suitable to edit multicopy genes. One such particular example corresponds to the mutagenesis of candidate-essential genes that requires the presence of an extra copy of the gene before gene disruption. This opens new insights to explore gene essentiality in bacteria and to limit off-target effects during systematic CRISPR-Cas9 based approaches.
基于 CRISPR/Cas(成簇规律间隔短回文重复序列和 CRISPR 相关核酸酶)的技术已经彻底改变了基因组工程。虽然它们在原核基因组编辑中的应用正在扩大,但仍存在一些局限性,例如可能的脱靶效应和设计限制。当在不同的基因座或靶向重复序列(例如,多拷贝基因或移动遗传元件)时,这些限制更加复杂。为了克服这些限制,我们设计了一种使用相同 sgRNA 和 CRISPR-Cas9 系统在不同基因座独立进行基因编辑的方法。
我们开发了一种两步程序,基于在感兴趣的基因拷贝附近通过同源重组引入“诱饵”DNA,然后诱导 CRISPR-Cas9 活性。引入靶向该“诱饵”DNA 的 CRISPR-Cas9 复合物的遗传工具会在感兴趣的拷贝附近诱导双链断裂。其通过同源重组修复可能导致回复或基因拷贝特异性编辑。这些事件的相对频率与基因编辑对细胞适应性的影响有关。在我们的研究中,我们使用该技术成功删除了链霉菌 ambofaciens 中两个异种沉默子 lsr2 基因的天然拷贝。我们观察到,其中一个基因是候选必需基因,因为只有在存在额外拷贝的情况下才能删除其天然基因座。
通过靶向“诱饵”DNA,我们设计了一种“通用”CRISPR-Cas9 工具包,可用于编辑不同的基因座。该 CRISPR-Cas9 系统的差异作用仅基于感兴趣的基因拷贝周围区域之间的特异性重组。这种方法适用于编辑多拷贝基因。一个特殊的例子是诱变候选必需基因,这需要在基因中断之前存在基因的额外拷贝。这为探索细菌中的基因必需性以及在基于 CRISPR-Cas9 的系统方法中限制脱靶效应提供了新的思路。