Brüggemann Holger, Poehlein Anja, Brzuszkiewicz Elzbieta, Scavenius Carsten, Enghild Jan J, Al-Zeer Munir A, Brinkmann Volker, Jensen Anders, Söderquist Bo
Department of Biomedicine, Aarhus University, Aarhus, Denmark.
Department of Genomic and Applied Microbiology, Institute of Microbiology and Genetics, University of Göttingen, Göttingen, Germany.
Front Microbiol. 2019 Mar 12;10:478. doi: 10.3389/fmicb.2019.00478. eCollection 2019.
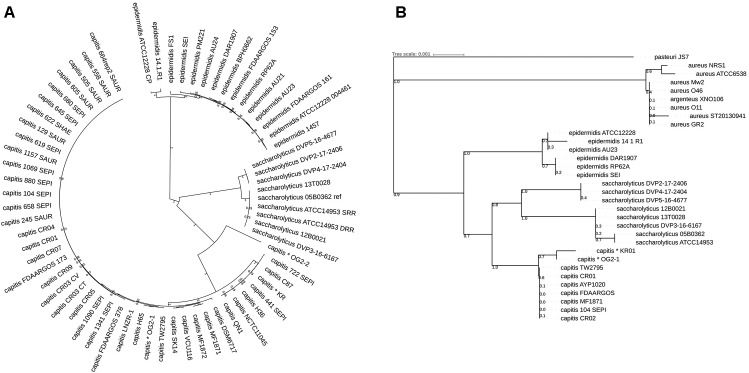
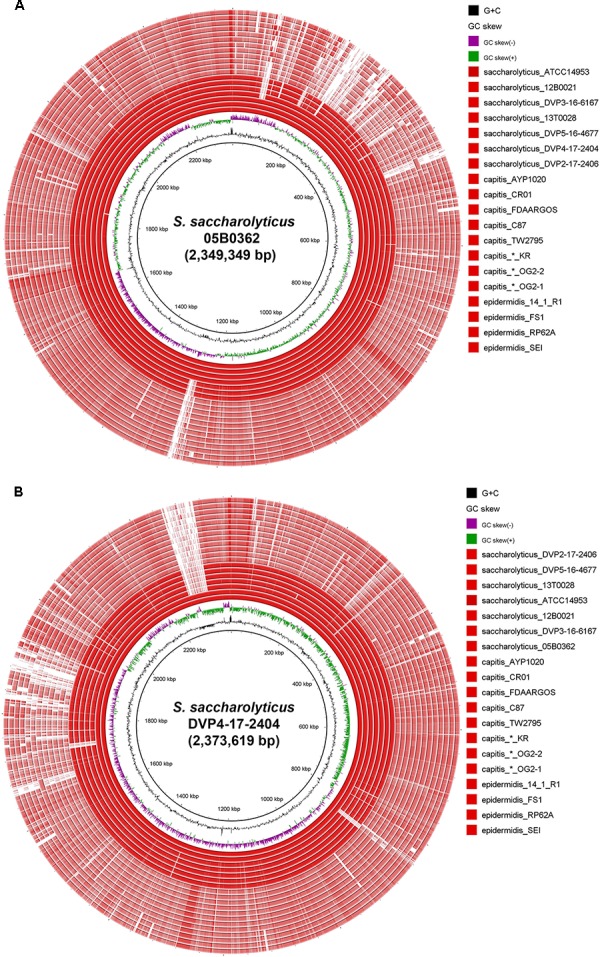
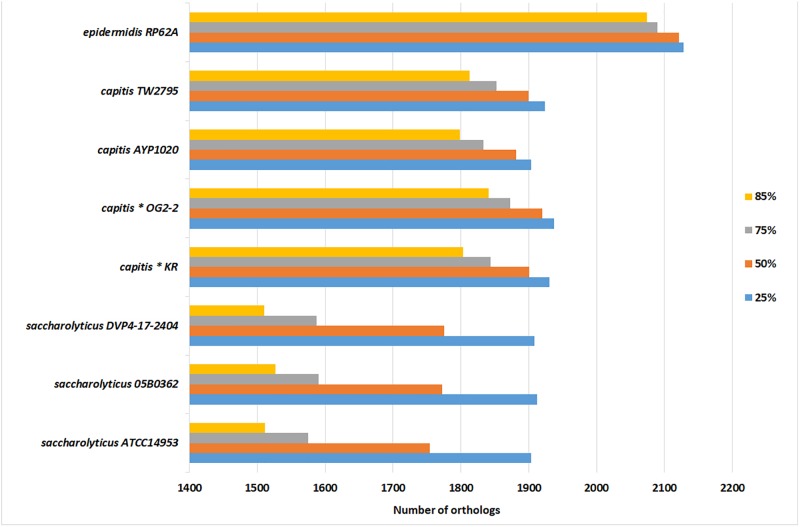
The slow-growing, anaerobic, coagulase-negative species is found on human skin and in clinical specimens but its pathogenic potential is unclear. Here, we investigated clinical isolates and sequenced the genomes of seven strains of . Phylogenomic analyses showed that the closest relative of is with an average nucleotide identity of 80%. Previously sequenced strains assigned to are misclassified and belong to . Based on single nucleotide polymorphisms of the core genome, the population of can be divided into two clades that also differ in a few larger genomic islands as part of the flexible genome. An unexpected feature of is extensive genome decay, with over 300 pseudogenes, indicating ongoing reductive evolution. Many genes of the core metabolism are not functional, rendering the species auxotrophic for several amino acids, which could explain its slow growth and need for fastidious growth conditions. Secreted proteins of were determined; they include stress response proteins such as heat and oxidative stress-related factors, as well as immunodominant staphylococcal surface antigens and enzymes that can degrade host tissue components. The strains secrete lipases and a hyaluronic acid lyase. Hyaluronidase as well as urease activities were detected in biochemical assays, with clade-specific differences. Our study revealed that has adapted its genome, possibly due to a recent change of habitat; moreover, the data imply that the species has tissue-invasive potential and might cause prosthetic joint infections.
这种生长缓慢、厌氧、凝固酶阴性的菌种存在于人体皮肤和临床标本中,但其致病潜力尚不清楚。在此,我们对临床分离株进行了研究,并对七株该菌种的基因组进行了测序。系统基因组分析表明,该菌种最接近的亲缘种是,平均核苷酸同一性为80%。先前归类为该菌种的已测序菌株分类错误,属于。基于核心基因组的单核苷酸多态性,该菌种的群体可分为两个进化枝,它们在一些较大的基因组岛(作为灵活基因组的一部分)上也存在差异。该菌种的一个意外特征是广泛的基因组退化,有超过300个假基因,表明正在进行简化进化。许多核心代谢基因无功能,使该菌种对几种氨基酸营养缺陷,这可以解释其生长缓慢以及对苛刻生长条件的需求。确定了该菌种的分泌蛋白;它们包括应激反应蛋白,如热和氧化应激相关因子,以及免疫显性葡萄球菌表面抗原和可降解宿主组织成分的酶。这些菌株分泌脂肪酶和一种透明质酸裂解酶。在生化分析中检测到透明质酸酶以及脲酶活性,存在进化枝特异性差异。我们的研究表明,该菌种可能由于最近栖息地的变化而使其基因组发生了适应性改变;此外,数据表明该菌种具有组织侵袭潜力,可能导致人工关节感染。