Department of Pediatrics, Erasmus University Medical Center, Rotterdam, Netherlands; Department of Clinical Genetics, Erasmus University Medical Center, Rotterdam, Netherlands; Center for Lysosomal and Metabolic Diseases, Erasmus University Medical Center, 3015 GE Rotterdam, Netherlands.
Department of Pediatrics, Erasmus University Medical Center, Rotterdam, Netherlands; Center for Lysosomal and Metabolic Diseases, Erasmus University Medical Center, 3015 GE Rotterdam, Netherlands.
EBioMedicine. 2019 May;43:553-561. doi: 10.1016/j.ebiom.2019.03.048. Epub 2019 Mar 25.
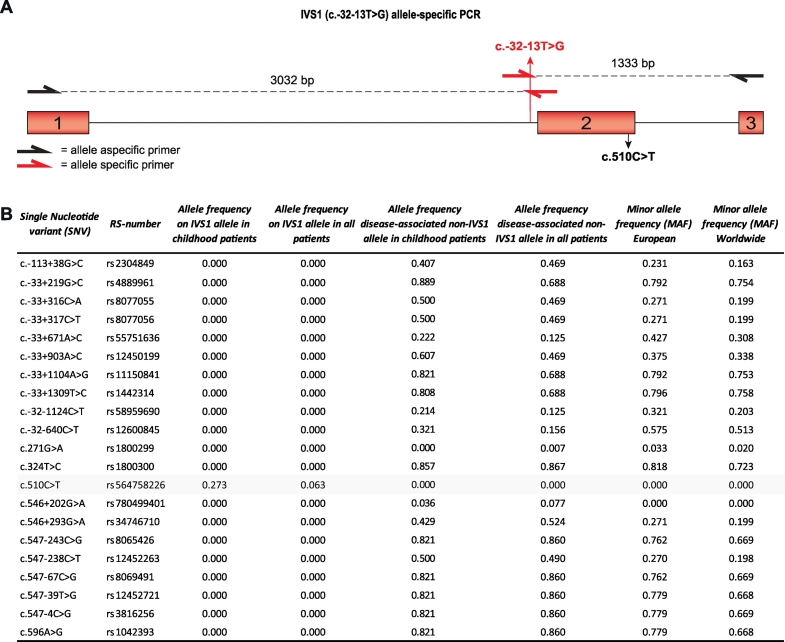
Neonatal screening for Pompe disease is complicated by difficulties in predicting symptom onset in patients with the common c.-32-13T>G (IVS1) variant/null (i.e. fully deleterious) acid α-glucosidase (GAA) genotype. This splicing variant occurs in 90% of Caucasian late onset patients, and is associated with a broad range of symptom onset.
We analyzed a cohort of 143 compound heterozygous and 10 homozygous IVS1 patients, and we assessed ages at symptom onset, the presence of cis-acting single nucleotide variants (SNVs), and performed splicing analysis and enzyme activity assays.
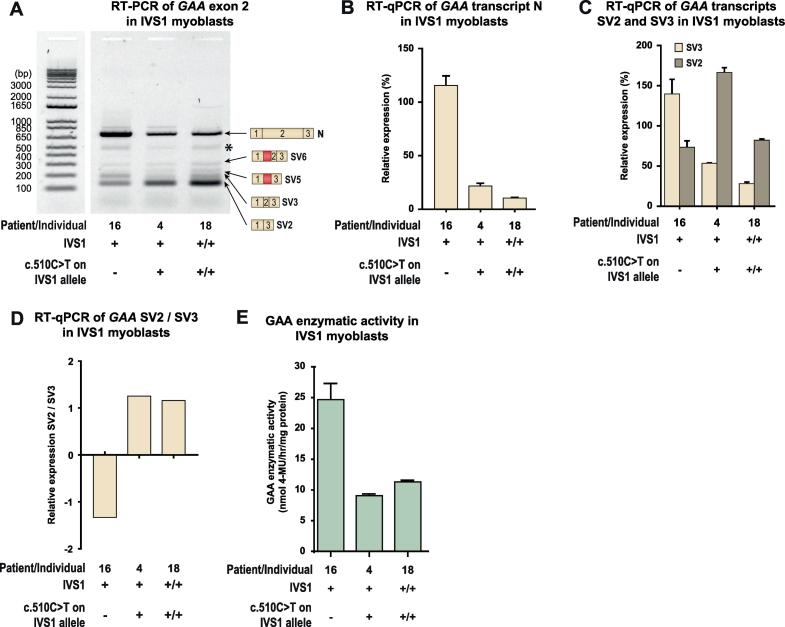
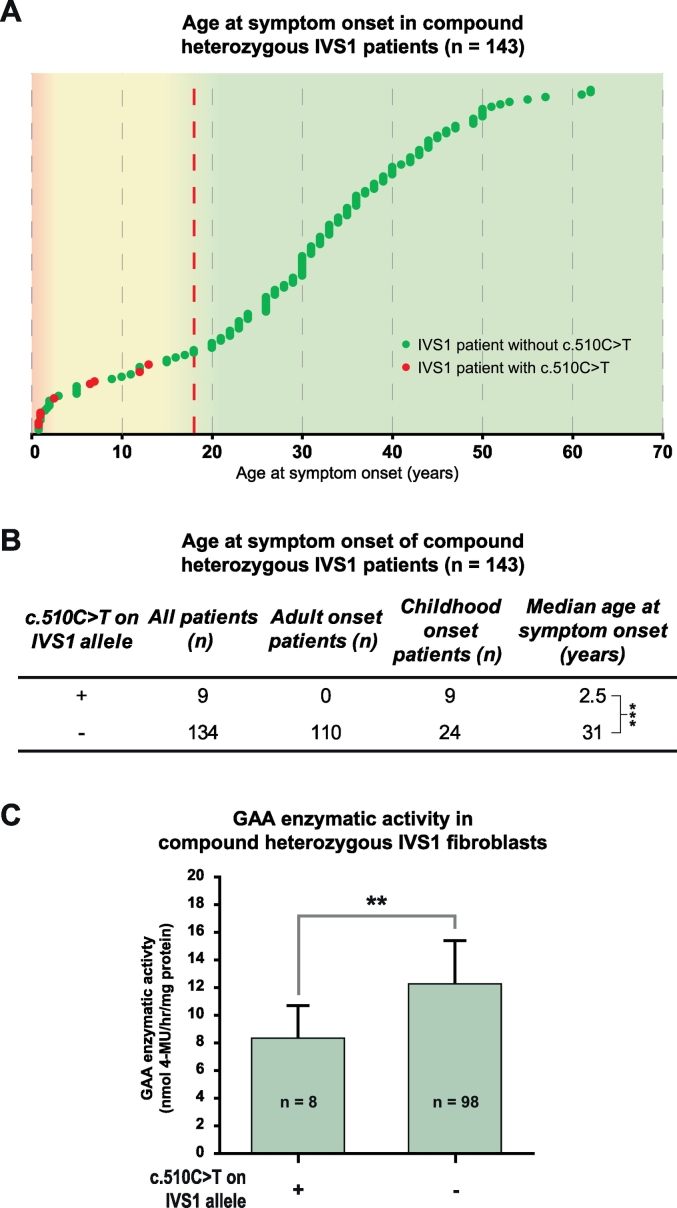
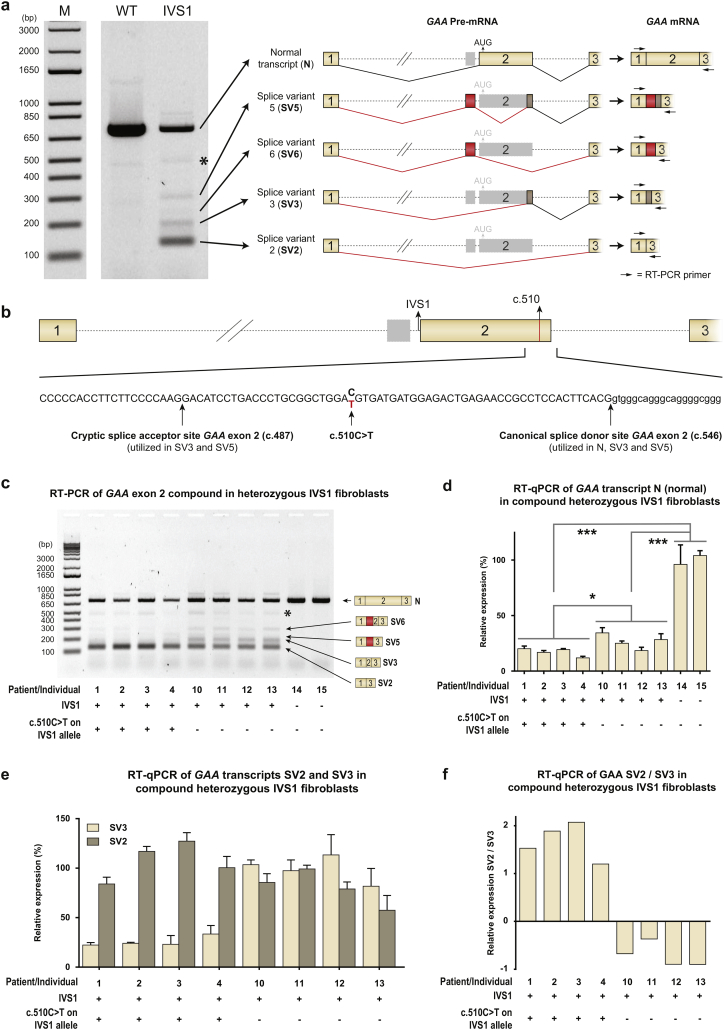
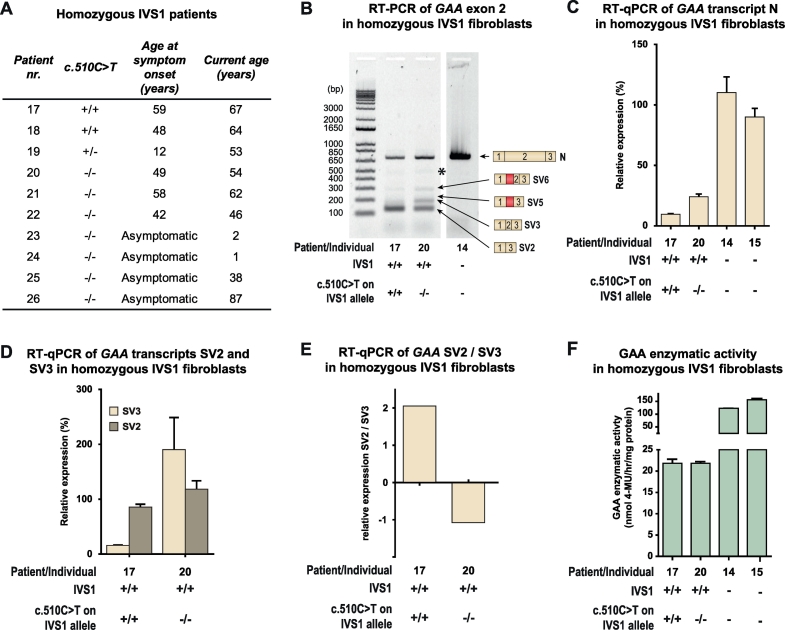
In compound heterozygous IVS1 patients, the synonymous variant c.510C>T was uniquely present on the IVS1 allele in 9/33 (27%) patients with childhood onset, but was absent from 110 patients with onset in adulthood. GAA enzyme activity was lower in fibroblasts from patients who contained c.510C>T than it was in patients without c.510C>T. By reducing the extent of leaky wild-type splicing, c.510C>T modulated aberrant splicing caused by the IVS1 variant. The deleterious effect of c.510C>T was also found in muscle cells, the main target cells in Pompe disease. In homozygous IVS1 patients, the c.510C>T variant was absent in 4/4 (100%) asymptomatic individuals and present in 3/6 (50%) symptomatic patients. In cells from homozygous IVS1 patients, c.510C>T caused reduced leaky wild-type splicing.
c.510C>T is a genetic modifier in compound heterozygous and homozygous IVS1 patients. This finding is important for neonatal screening programs for Pompe disease. FUND: This work was funded by grants from Sophia Children's Hospital Foundation (SSWO, grant S17-32) and Metakids (2016-063).
庞贝病的新生儿筛查受到了挑战,因为难以预测常见 c.-32-13T>G(IVS1)变异/缺失(即完全有害)酸性α-葡萄糖苷酶(GAA)基因型患者的症状发作时间。这种剪接变异发生在 90%的高加索晚发型患者中,与广泛的症状发作时间相关。
我们分析了 143 例复合杂合子和 10 例纯合子 IVS1 患者的队列,评估了症状发作的年龄、顺式作用单核苷酸变异(SNV)的存在情况,并进行了剪接分析和酶活性测定。
在复合杂合子 IVS1 患者中,同义变异 c.510C>T 仅存在于 9/33(27%)儿童期发病患者的 IVS1 等位基因上,而在 110 例成年期发病患者中不存在。c.510C>T 存在的患者的成纤维细胞中的 GAA 酶活性低于不存在 c.510C>T 的患者。通过减少野生型漏读剪接的程度,c.510C>T 调节了由 IVS1 变异引起的异常剪接。在肌肉细胞中也发现了 c.510C>T 的有害作用,肌肉细胞是庞贝病的主要靶细胞。在纯合子 IVS1 患者中,4/4(100%)无症状个体中不存在 c.510C>T 变异,而 3/6(50%)症状性患者中存在 c.510C>T 变异。在纯合子 IVS1 患者的细胞中,c.510C>T 导致野生型漏读剪接减少。
c.510C>T 是复合杂合子和纯合子 IVS1 患者的遗传修饰因子。这一发现对庞贝病的新生儿筛查计划具有重要意义。
本工作得到 Sophia 儿童医院基金会(SSWO,授予 S17-32)和 Metakids(2016-063)的资助。