Vaccine and Infectious Disease Division and Statistical Center for HIV/AIDS Research and Prevention, Fred Hutchinson Cancer Research Center, Seattle, Washington, United States of America.
Department of Biostatistics and Bioinformatics, Rollins School of Public Health, Emory University, Atlanta, Georgia, United States of America.
PLoS Comput Biol. 2019 Apr 1;15(4):e1006952. doi: 10.1371/journal.pcbi.1006952. eCollection 2019 Apr.
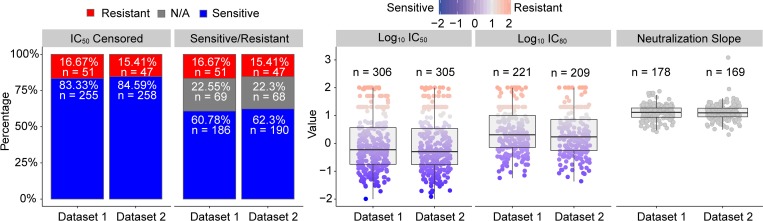
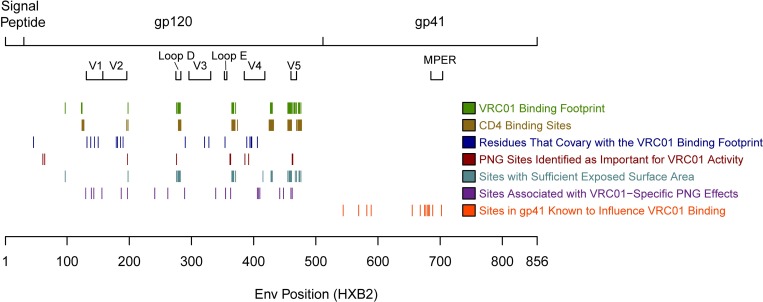
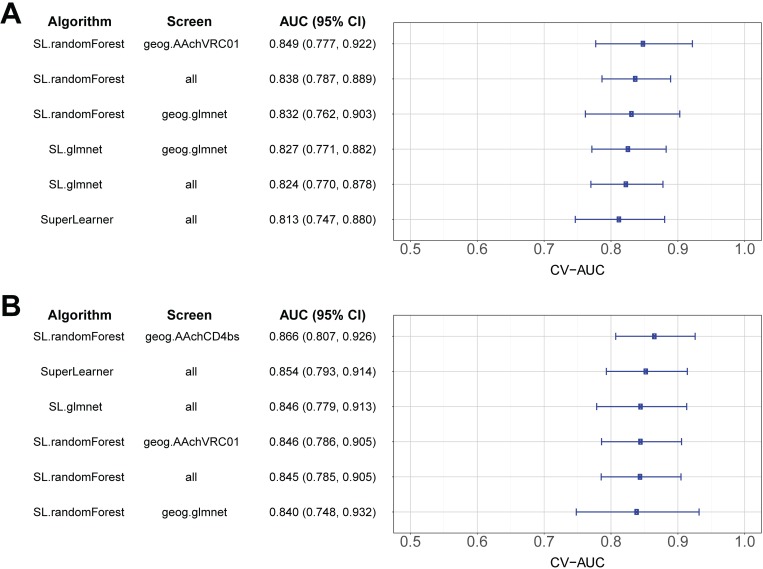
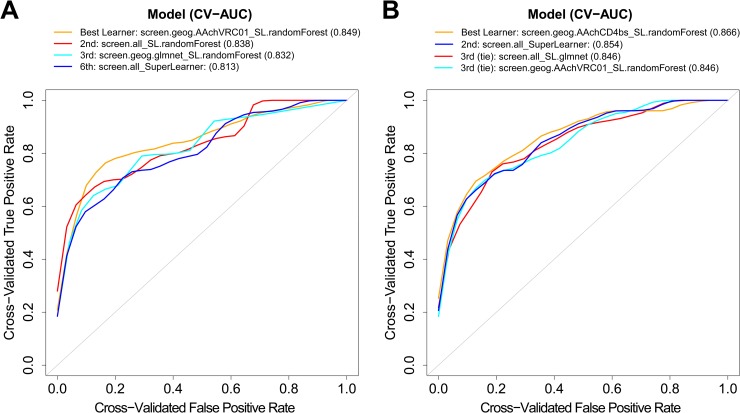
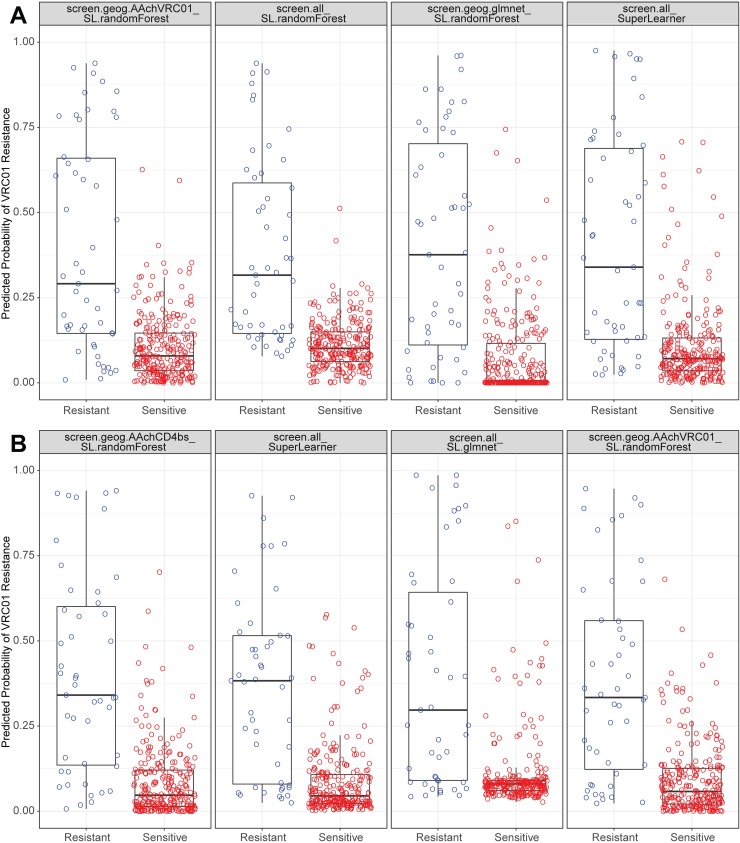
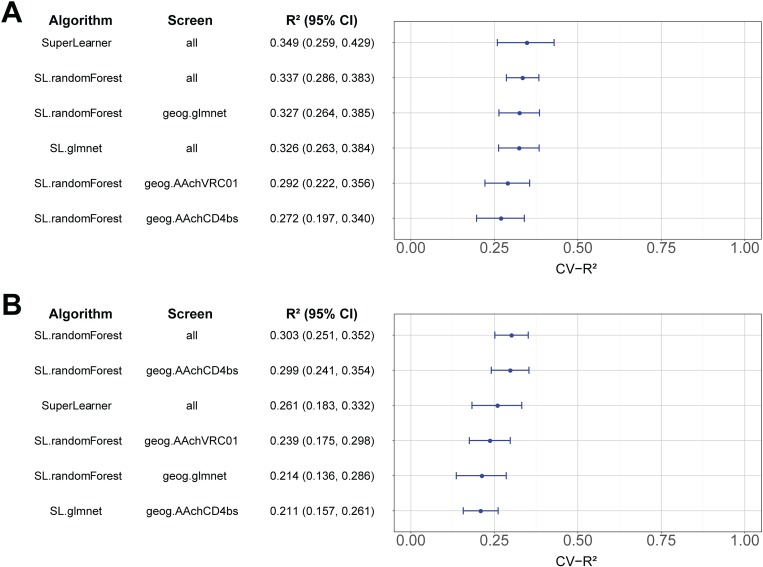
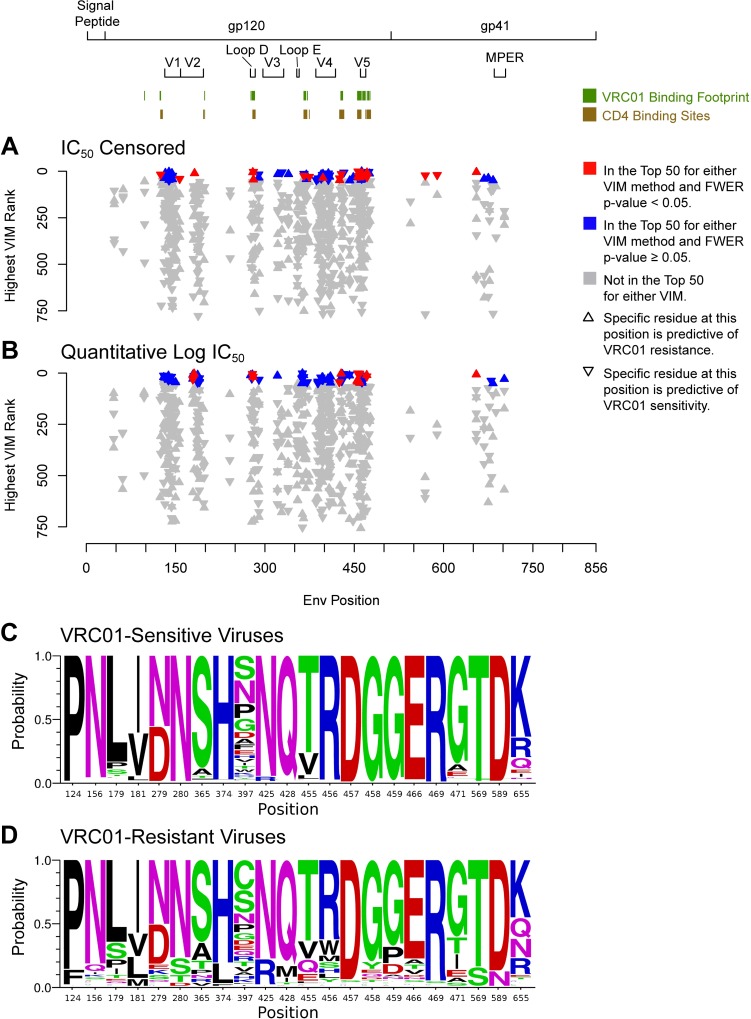
The broadly neutralizing antibody (bnAb) VRC01 is being evaluated for its efficacy to prevent HIV-1 infection in the Antibody Mediated Prevention (AMP) trials. A secondary objective of AMP utilizes sieve analysis to investigate how VRC01 prevention efficacy (PE) varies with HIV-1 envelope (Env) amino acid (AA) sequence features. An exhaustive analysis that tests how PE depends on every AA feature with sufficient variation would have low statistical power. To design an adequately powered primary sieve analysis for AMP, we modeled VRC01 neutralization as a function of Env AA sequence features of 611 HIV-1 gp160 pseudoviruses from the CATNAP database, with objectives: (1) to develop models that best predict the neutralization readouts; and (2) to rank AA features by their predictive importance with classification and regression methods. The dataset was split in half, and machine learning algorithms were applied to each half, each analyzed separately using cross-validation and hold-out validation. We selected Super Learner, a nonparametric ensemble-based cross-validated learning method, for advancement to the primary sieve analysis. This method predicted the dichotomous resistance outcome of whether the IC50 neutralization titer of VRC01 for a given Env pseudovirus is right-censored (indicating resistance) with an average validated AUC of 0.868 across the two hold-out datasets. Quantitative log IC50 was predicted with an average validated R2 of 0.355. Features predicting neutralization sensitivity or resistance included 26 surface-accessible residues in the VRC01 and CD4 binding footprints, the length of gp120, the length of Env, the number of cysteines in gp120, the number of cysteines in Env, and 4 potential N-linked glycosylation sites; the top features will be advanced to the primary sieve analysis. This modeling framework may also inform the study of VRC01 in the treatment of HIV-infected persons.
广谱中和抗体 (bnAb) VRC01 正在抗体制动预防 (AMP) 试验中评估其预防 HIV-1 感染的功效。AMP 的次要目标是利用筛析分析研究 VRC01 预防功效 (PE) 如何随 HIV-1 包膜 (Env) 氨基酸 (AA) 序列特征而变化。对每一个具有足够变化的 AA 特征进行测试以了解 PE 如何变化的详尽分析将具有较低的统计能力。为了设计 AMP 的一个充分有力的主要筛析分析,我们将 VRC01 的中和作用建模为 CATNAP 数据库中 611 个 HIV-1 gp160 假病毒的 Env AA 序列特征的函数,目标是:(1) 开发最能预测中和反应的模型;(2) 利用分类和回归方法根据 AA 特征的预测重要性对其进行排序。数据集被分为两半,机器学习算法应用于每一半,通过交叉验证和保留验证分别进行分析。我们选择了 Super Learner,一种非参数基于集合的交叉验证学习方法,用于推进主要筛析分析。该方法通过平均验证 AUC 为 0.868 来预测 VRC01 对特定 Env 假病毒的 IC50 中和滴度是否右删失(表示耐药)的二项耐药结果,这在两个保留数据集上均有效。定量 log IC50 的预测平均验证 R2 为 0.355。预测中和敏感性或耐药性的特征包括 VRC01 和 CD4 结合足迹中的 26 个表面可及残基、gp120 的长度、Env 的长度、gp120 中的半胱氨酸数、Env 中的半胱氨酸数和 4 个潜在的 N-糖基化位点;顶级特征将被推进主要筛析分析。该建模框架还可以为 VRC01 在治疗 HIV 感染者中的研究提供信息。