Department of Neurology, University Hospital Würzburg, Josef-Schneider-Str. 11, 97080, Würzburg, Germany.
University Hospital Würzburg, Institute for Clinical Neurobiology, Versbacher Str. 5, 97078, Würzburg, Germany.
J Neuroinflammation. 2019 Apr 5;16(1):73. doi: 10.1186/s12974-019-1462-z.
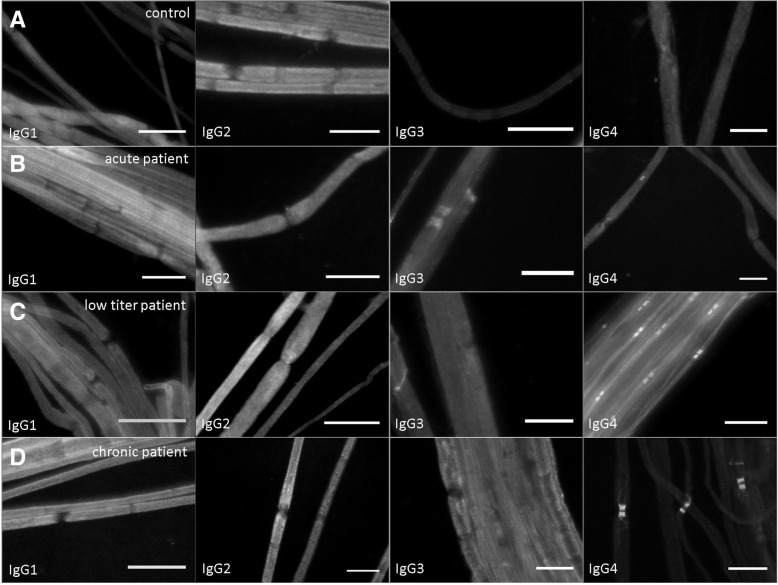
Autoantibodies against the paranodal protein contactin-1 have recently been described in patients with severe acute-onset autoimmune neuropathies and mainly belong to the IgG4 subclass that does not activate complement. IgG3 anti-contactin-1 autoantibodies are rare, but have been detected during the acute onset of disease in some cases. There is evidence that anti-contactin-1 prevents adhesive interaction, and chronic exposure to anti-contactin-1 IgG4 leads to structural changes at the nodes accompanied by neuropathic symptoms. However, the pathomechanism of acute onset of disease and the pathogenic role of IgG3 anti-contactin-1 is largely unknown.
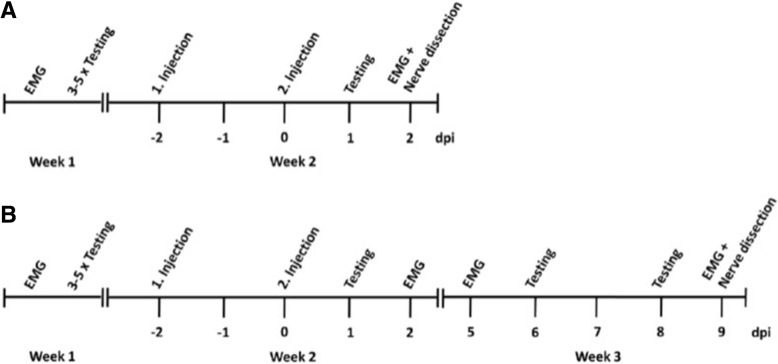
In the present study, we aimed to model acute autoantibody exposure by intraneural injection of IgG of patients with anti-contacin-1 autoantibodies to Lewis rats. Patient IgG obtained during acute onset of disease (IgG3 predominant) and IgG from the chronic phase of disease (IgG4 predominant) were studied in comparison.
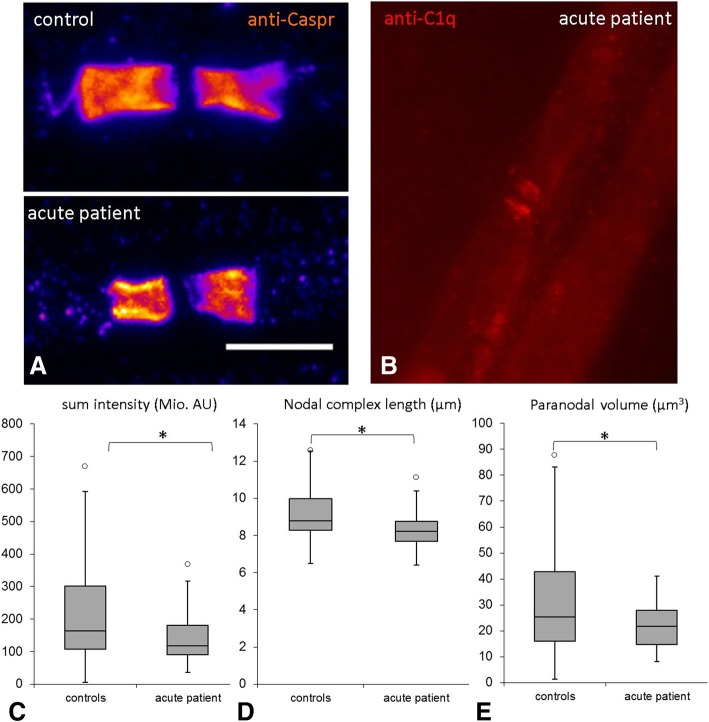
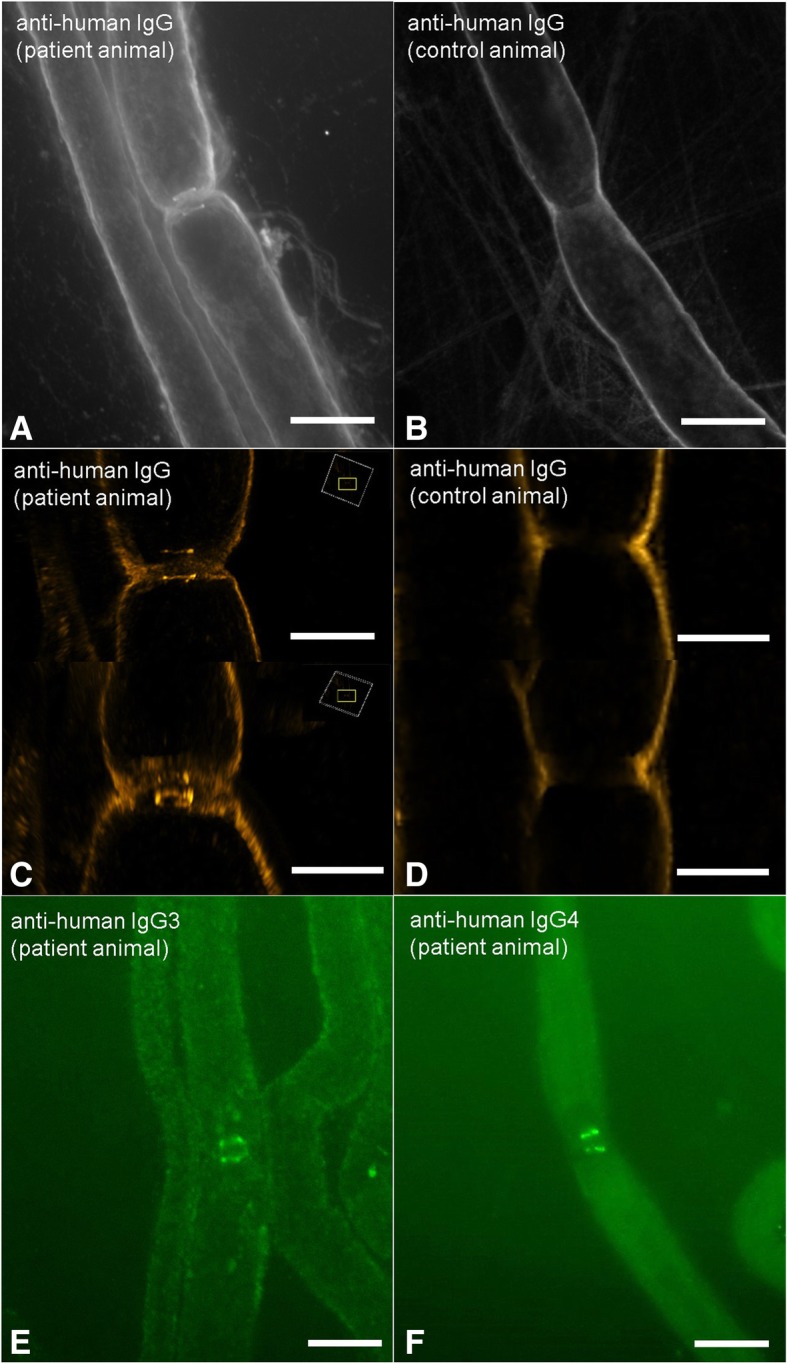
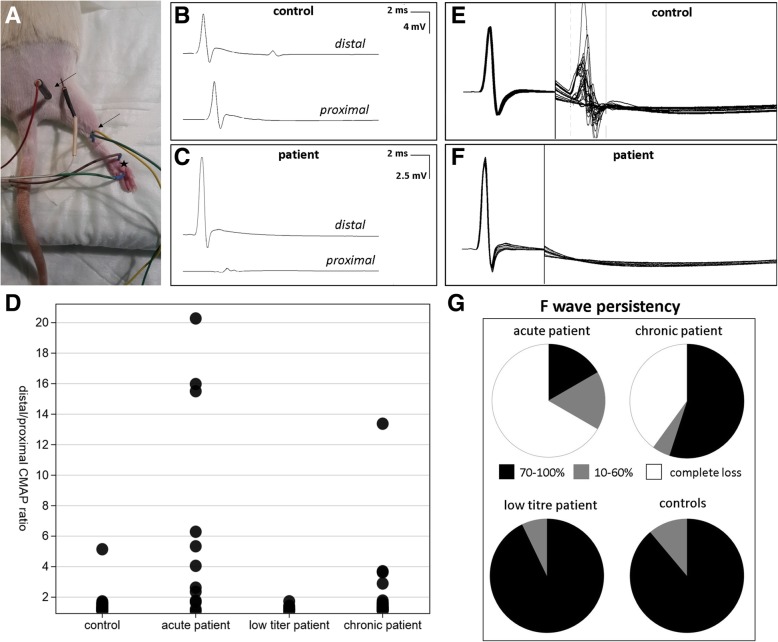
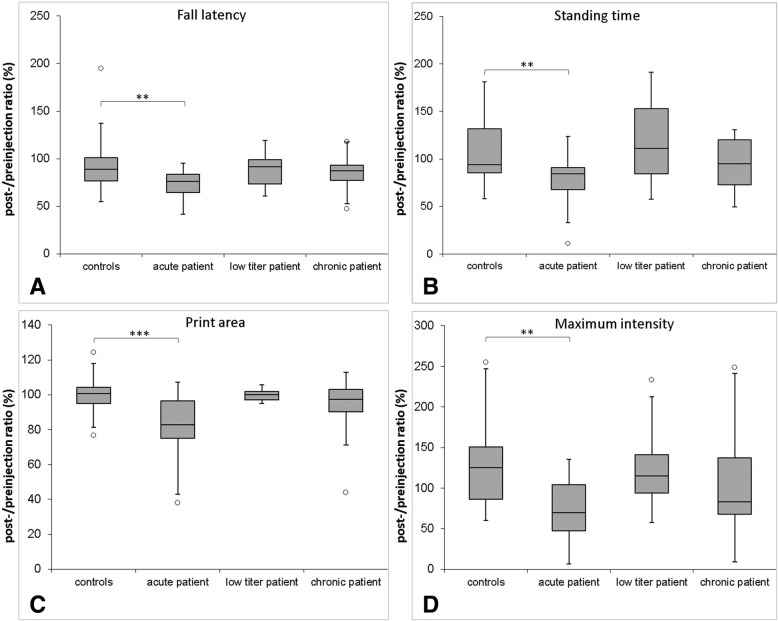
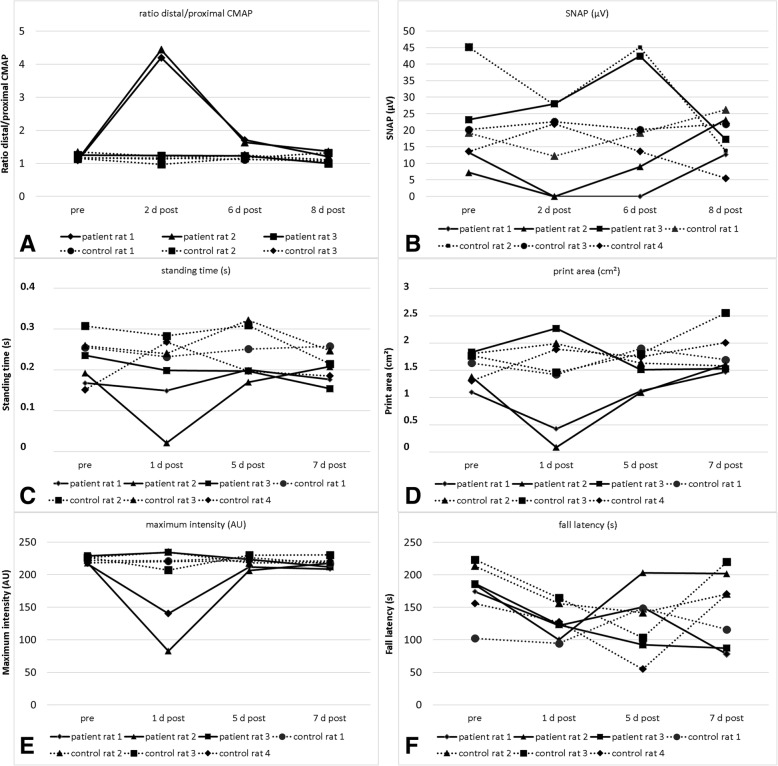
Conduction blocks were measured in rats injected with the "acute" IgG more often than after injection of "chronic" IgG (83.3% versus 35%) and proved to be reversible within a week after injection. Impaired nerve conduction was accompanied by motor deficits in rats after injection of the "acute" IgG but only minor structural changes of the nodes. Paranodal complement deposition was detected after injection of the "acute IgG". We did not detect any inflammatory infiltrates, arguing against an inflammatory cascade as cause of damage to the nerve. We also did not observe dispersion of paranodal proteins or sodium channels to the juxtaparanodes as seen in patients after chronic exposure to anti-contactin-1.
Our data suggest that anti-contactin-1 IgG3 induces an acute conduction block that is most probably mediated by autoantibody binding and subsequent complement deposition and may account for acute onset of disease in these patients. This supports the notion of anti-contactin-1-associated neuropathy as a paranodopathy with the nodes of Ranvier as the site of pathogenesis.
最近在严重急性自身免疫性神经病患者中描述了针对神经节旁蛋白接触蛋白-1 的自身抗体,这些自身抗体主要属于不激活补体的 IgG4 亚类。IgG3 抗接触蛋白-1 自身抗体很少见,但在某些情况下已在疾病急性发作时检测到。有证据表明,抗接触蛋白-1 可阻止黏附相互作用,而慢性暴露于抗接触蛋白-1 IgG4 会导致伴随神经病症状的神经节旁结构发生变化。然而,疾病急性发作的发病机制和 IgG3 抗接触蛋白-1 的致病作用在很大程度上仍是未知的。
在本研究中,我们旨在通过向 Lewis 大鼠的神经内注射抗接触蛋白-1 自身抗体的患者 IgG 来模拟急性自身抗体暴露。研究了疾病急性发作期(以 IgG3 为主)和疾病慢性期(以 IgG4 为主)获得的患者 IgG。
与注射“慢性”IgG 相比,注射“急性”IgG 的大鼠更常出现传导阻滞(83.3%比 35%),并且在注射后一周内可恢复。在注射“急性”IgG 后,大鼠的神经传导受损伴有运动功能障碍,但仅观察到神经节旁的轻微结构变化。注射“急性”IgG 后检测到神经节旁补体沉积。我们未发现任何炎症浸润,这表明神经损伤不是炎症级联反应的结果。我们也未观察到与慢性接触蛋白-1 暴露后患者一样的轴索旁蛋白或钠通道向节旁区的离散。
我们的数据表明,抗接触蛋白-1 IgG3 诱导急性传导阻滞,这很可能是通过自身抗体结合和随后的补体沉积介导的,可能是这些患者疾病急性发作的原因。这支持了接触蛋白-1 相关神经病是一种以Ranvier 神经节为发病部位的神经节旁病的观点。