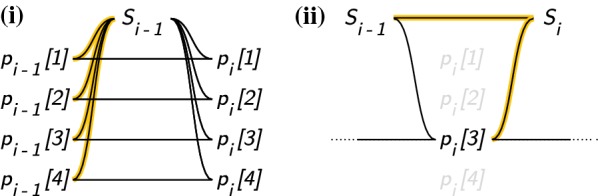Rosen Yohei M, Paten Benedict J
1UCSC Genomics Institute, 1156 High St, Santa Cruz, CA 95064 USA.
2NYU School of Medicine, 550 First Ave, New York, NY 10016 USA.
Algorithms Mol Biol. 2019 Apr 2;14:11. doi: 10.1186/s13015-019-0144-9. eCollection 2019.
Hidden Markov models of haplotype inheritance such as the Li and Stephens model allow for computationally tractable probability calculations using the forward algorithm as long as the representative reference panel used in the model is sufficiently small. Specifically, the monoploid Li and Stephens model and its variants are linear in reference panel size unless heuristic approximations are used. However, sequencing projects numbering in the thousands to hundreds of thousands of individuals are underway, and others numbering in the millions are anticipated.
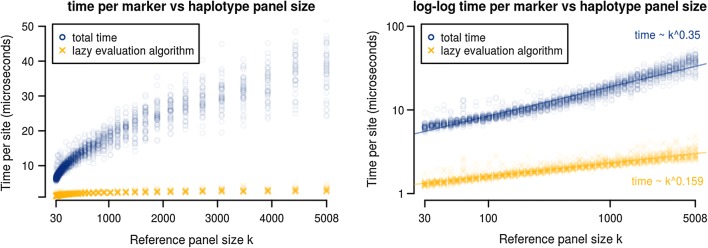
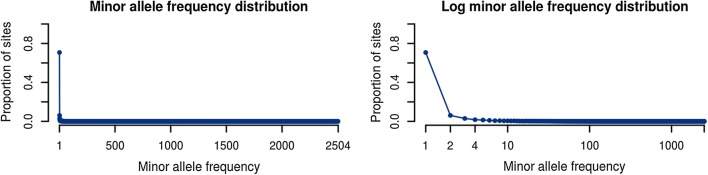
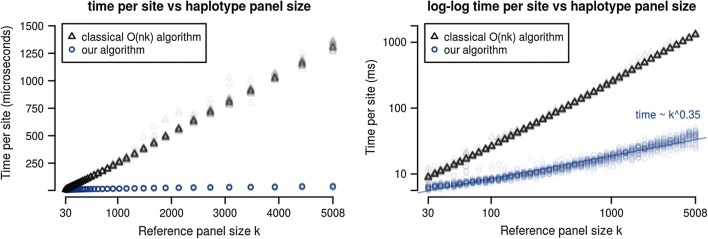
To make the forward algorithm for the haploid Li and Stephens model computationally tractable for these datasets, we have created a numerically exact version of the algorithm with observed average case sublinear runtime with respect to reference panel size when tested against the 1000 Genomes dataset.
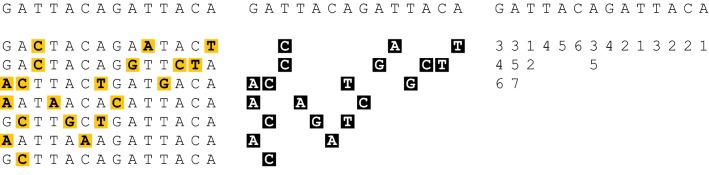
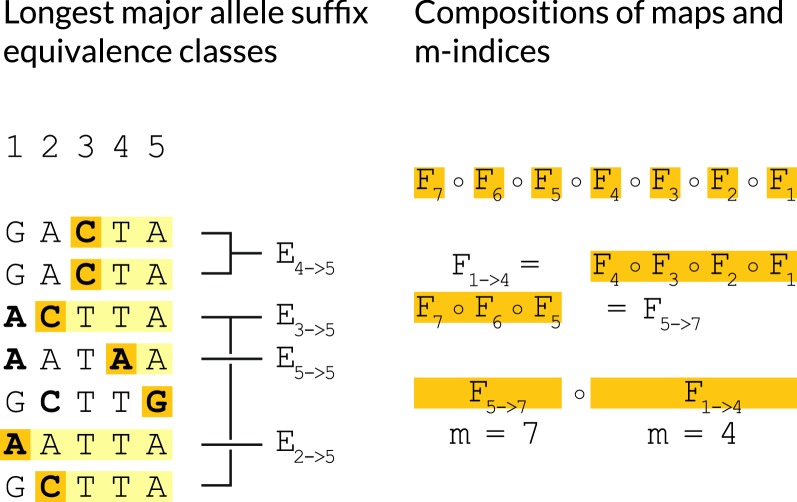
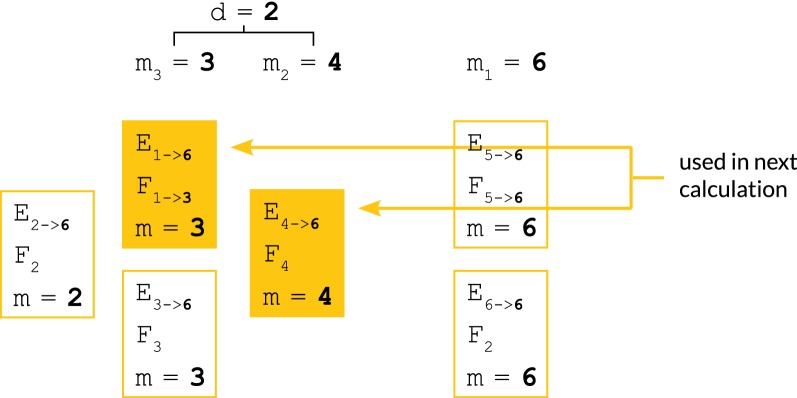
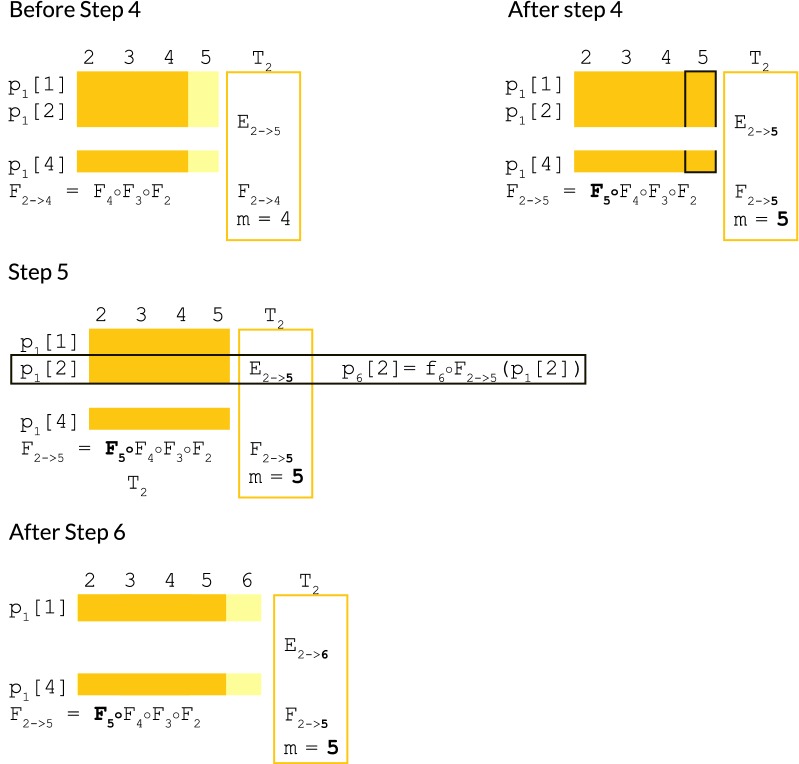
We show a forward algorithm which avoids any tradeoff between runtime and model complexity. Our algorithm makes use of two general strategies which might be applicable to improving the time complexity of other future sequence analysis algorithms: sparse dynamic programming matrices and lazy evaluation.
单倍型遗传的隐马尔可夫模型,如李和斯蒂芬斯模型,只要模型中使用的代表性参考面板足够小,就可以使用前向算法进行计算上易于处理的概率计算。具体而言,除非使用启发式近似,否则单倍型李和斯蒂芬斯模型及其变体在参考面板大小方面是线性的。然而,目前正在进行的测序项目涉及数千到数十万个体,预计还有数百万个体的测序项目。
为了使单倍型李和斯蒂芬斯模型的前向算法在这些数据集上计算上易于处理,我们创建了该算法的数值精确版本,在针对千人基因组数据集进行测试时,相对于参考面板大小,观察到平均情况的亚线性运行时间。
我们展示了一种前向算法,该算法避免了运行时间和模型复杂性之间的任何权衡。我们的算法利用了两种可能适用于提高未来其他序列分析算法时间复杂度的通用策略:稀疏动态规划矩阵和惰性求值。