Heng Ling Zhi, Kennedy Joanna, Smithson Sarah, Newbury-Ecob Ruth, Churchill Amanda
Department of Paediatric Ophthalmology, Bristol Eye Hospital, Bristol, UK.
Clinical Genetics, University Hospitals Bristol, Bristol, UK.
BMJ Open Ophthalmol. 2019 Feb 16;4(1):e000234. doi: 10.1136/bmjophth-2018-000234. eCollection 2019.
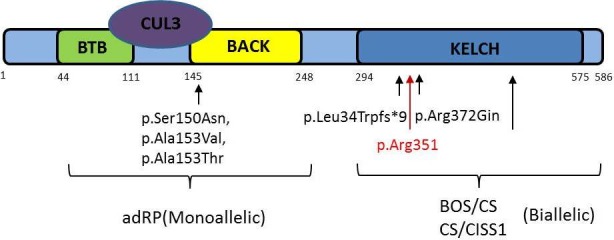
The ubiquitin-proteasome system pathway has been recognised as a crucial cellular mechanism for the proper function of photoreceptor cells. In particular, ubiquitin ligases (E3s) recognise and ubiquitinate specific proteins for degradation. The KLHL7 protein (a BTB-Kelch protein) has been found to play an important role in this process. There have been several reports that heterozygous mutations in the gene in adults are responsible for a rare cause of late-onset autosomal dominant retinitis pigmentosa with preservation of central vision and homozygous mutations in two young children, with Crisponi syndrome (CS)/cold-induced sweating syndrome type 1, result in a recessive form of early-onset peripheral retinal dystrophy type changes. The majority of children do not survive through to adulthood. The objective of this study is to report the visual symptoms and signs of two young adults clinically diagnosed with overlapping BOS/Cisproni syndrome, expanding the phenotypic presentation of KLHL7 gene mutations.
This is a case report of the ophthalmic findings of two siblings with biallelic KLHL7 gene mutations. Siblings born to a non-consanguineous family and diagnosed with the overlapping clinical phenotype of Bohring-Opitz and and confirmed biallelic KLHL 7 gene mutation by whole exome sequencing were identified. Ophthlamic history and fundal examination was performed and analysed.
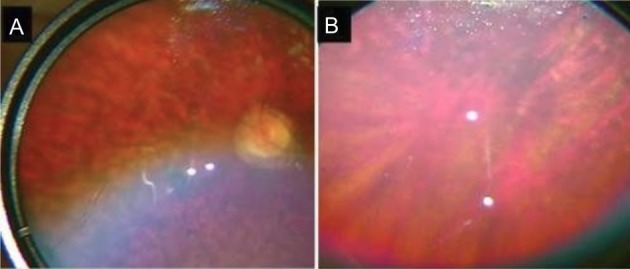
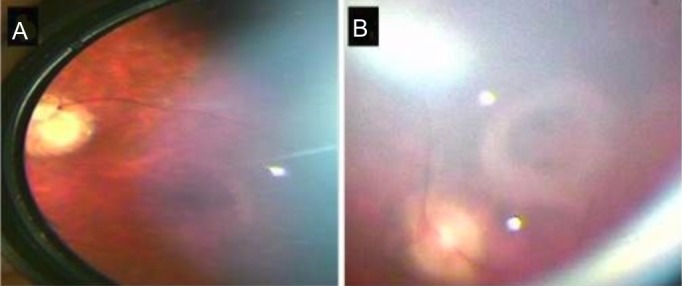
Both patients had similar retinal findings. The fundus shows confluent hypopigmented/pale yellow lesions in the mid-periphery. The optic disc appears to be pale with a ring of atrophy and vessels appear attenuated. The macular of the younger patient shows a depigmented area around the fovea giving a bull's-eye appearance while the older sibling shows a fibrotic ring around the fovea suggesting a more advanced pathology.
This paper expands the retinal phenotype to include a distinctive maculopathy in a recently described homozygous mutation in the gene in two young adults presenting with features that overlap the Bohring-Opitz syndrome and CS.
泛素 - 蛋白酶体系统途径已被公认为是光感受器细胞正常功能的关键细胞机制。特别是,泛素连接酶(E3s)识别并泛素化特定蛋白质以便降解。已发现KLHL7蛋白(一种BTB - Kelch蛋白)在此过程中起重要作用。有几份报告指出,成人该基因的杂合突变是导致罕见的晚发性常染色体显性视网膜色素变性且中心视力保留的原因,而两名幼儿的纯合突变与克里斯波尼综合征(CS)/ 1型冷诱导出汗综合征有关,会导致隐性形式的早发性周边视网膜营养不良类型改变。大多数儿童无法存活至成年。本研究的目的是报告两名临床诊断为重叠型BOS / 克里斯波尼综合征的年轻成年人的视觉症状和体征,以扩展KLHL7基因突变的表型表现。
这是一份关于两名具有双等位基因KLHL7基因突变的兄弟姐妹眼科检查结果的病例报告。通过全外显子测序鉴定出一对非近亲家庭出生的兄弟姐妹,他们被诊断为具有重叠的博林 - 奥皮茨临床表型,并确认存在双等位基因KLHL 7基因突变。进行并分析了眼科病史和眼底检查。
两名患者的视网膜表现相似。眼底显示中周边部有融合的色素减退/浅黄色病变。视盘看起来苍白,有萎缩环,血管变细。较年轻患者的黄斑在中央凹周围显示色素脱失区,呈靶心外观,而年长的兄弟姐妹在中央凹周围显示纤维化环,提示病理改变更严重。
本文扩展了视网膜表型,在两名表现出与博林 - 奥皮茨综合征和CS重叠特征的年轻成年人中,该基因最近描述的纯合突变包括一种独特的黄斑病变。