Departments of Bioengineering and Mechanical and Aerospace Engineering, University of California, San Diego, 9500 Gilman Dr, La Jolla, CA, 92093, USA.
Department of Bioengineering and San Diego Supercomputer Center, University of California, San Diego, 9500 Gilman Dr, La Jolla, CA, 92093, USA.
BMC Bioinformatics. 2019 Apr 27;20(1):212. doi: 10.1186/s12859-019-2746-0.
Community detection algorithms are fundamental tools to uncover important features in networks. There are several studies focused on social networks but only a few deal with biological networks. Directly or indirectly, most of the methods maximize modularity, a measure of the density of links within communities as compared to links between communities.
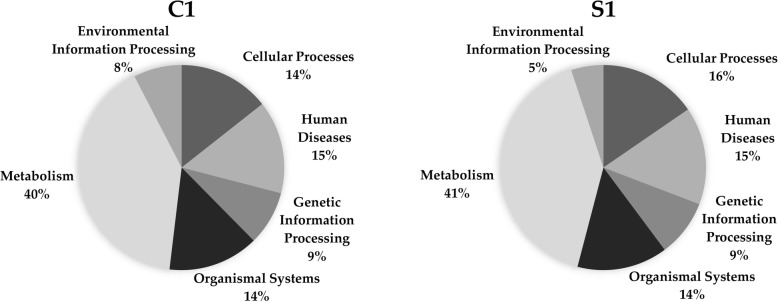
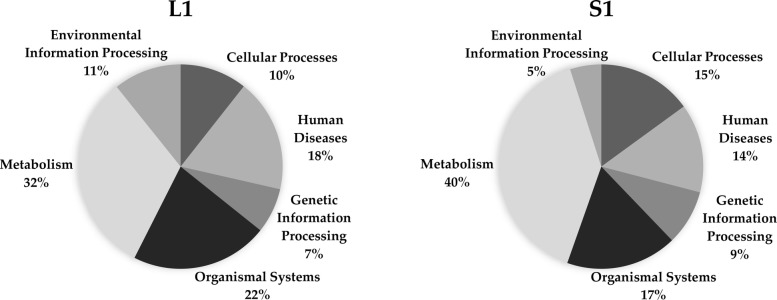
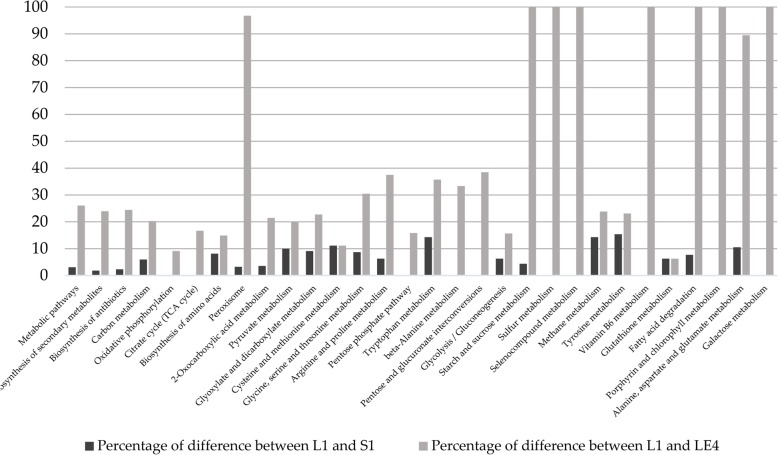
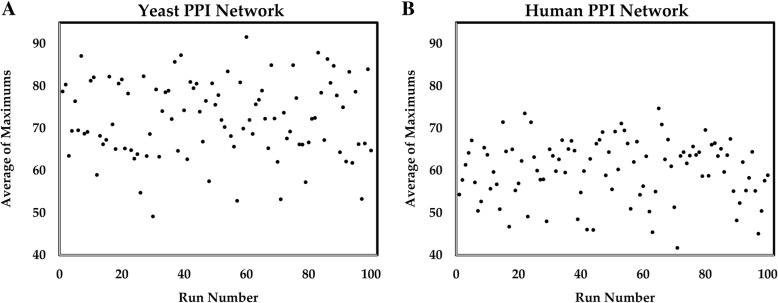
Here we analyze six different community detection algorithms, namely, Combo, Conclude, Fast Greedy, Leading Eigen, Louvain and Spinglass, on two important biological networks to find their communities and evaluate the results in terms of topological and functional features through Kyoto Encyclopedia of Genes and Genomes pathway and Gene Ontology term enrichment analysis. At a high level, the main assessment criteria are 1) appropriate community size (neither too small nor too large), 2) representation within the community of only one or two broad biological functions, 3) most genes from the network belonging to a pathway should also belong to only one or two communities, and 4) performance speed. The first network in this study is a network of Protein-Protein Interactions (PPI) in Saccharomyces cerevisiae (Yeast) with 6532 nodes and 229,696 edges and the second is a network of PPI in Homo sapiens (Human) with 20,644 nodes and 241,008 edges. All six methods perform well, i.e., find reasonably sized and biologically interpretable communities, for the Yeast PPI network but the Conclude method does not find reasonably sized communities for the Human PPI network. Louvain method maximizes modularity by using an agglomerative approach, and is the fastest method for community detection. For the Yeast PPI network, the results of Spinglass method are most similar to the results of Louvain method with regard to the size of communities and core pathways they identify, whereas for the Human PPI network, Combo and Spinglass methods yield the most similar results, with Louvain being the next closest.
For Yeast and Human PPI networks, Louvain method is likely the best method to find communities in terms of detecting known core pathways in a reasonable time.
社区检测算法是揭示网络重要特征的基本工具。有许多针对社交网络的研究,但只有少数涉及生物网络。大多数方法直接或间接地最大化模块性,这是社区内链接密度与社区间链接密度的度量。
在这里,我们分析了六种不同的社区检测算法,即 Combo、Conclude、Fast Greedy、Leading Eigen、Louvain 和 Spinglass,在两个重要的生物网络上寻找它们的社区,并通过京都基因与基因组百科全书通路和基因本体论术语富集分析来评估它们在拓扑和功能特征方面的结果。在较高的水平上,主要评估标准是 1)适当的社区大小(既不大也不小),2)社区内只代表一两个广泛的生物学功能,3)网络中的大多数基因都应该只属于一个或两个社区,4)性能速度。本研究中的第一个网络是酿酒酵母(酵母)的蛋白质-蛋白质相互作用(PPI)网络,有 6532 个节点和 229696 个边,第二个网络是人类的 PPI 网络,有 20644 个节点和 241008 个边。所有六种方法都表现良好,即找到合理大小和具有生物学可解释性的社区,对于酵母 PPI 网络,但 Conclude 方法对于人类 PPI 网络没有找到合理大小的社区。Louvain 方法通过聚合方法最大化模块性,是社区检测最快的方法。对于酵母 PPI 网络,Spinglass 方法的结果在社区的大小和它确定的核心通路方面与 Louvain 方法最相似,而对于人类 PPI 网络,Combo 和 Spinglass 方法的结果最相似,其次是 Louvain 方法。
对于酵母和人类 PPI 网络,Louvain 方法可能是在合理的时间内找到社区的最佳方法,以检测已知的核心通路。