Department of Pharmacology, Pharmacy College, Xinjiang Medical University, Ürümqi, Xinjiang Uyghur Autonomous Region 830001, P.R. China.
Department of Internal Medicine, Traditional Medical College, Xinjiang Medical University, Ürümqi, Xinjiang Uyghur Autonomous Region 830001, P.R. China.
Mol Med Rep. 2019 Jun;19(6):4697-4710. doi: 10.3892/mmr.2019.10165. Epub 2019 Apr 15.
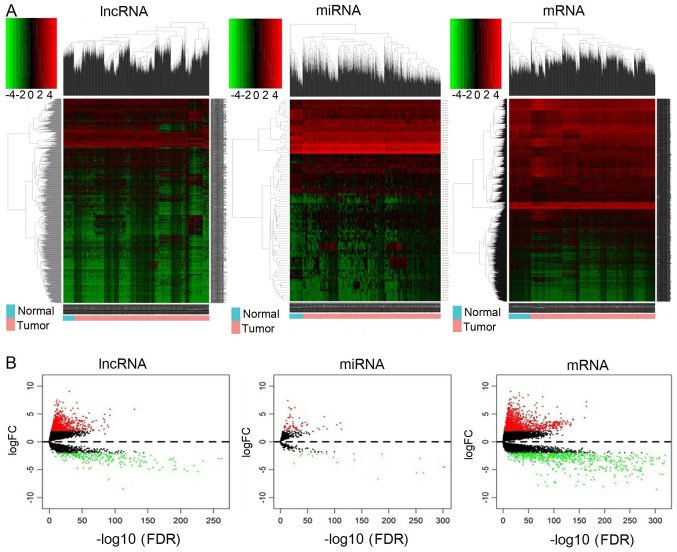
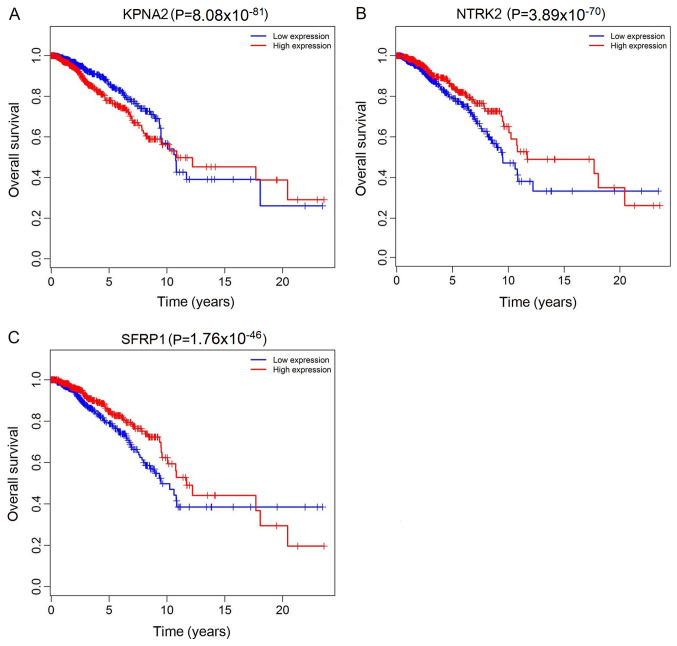
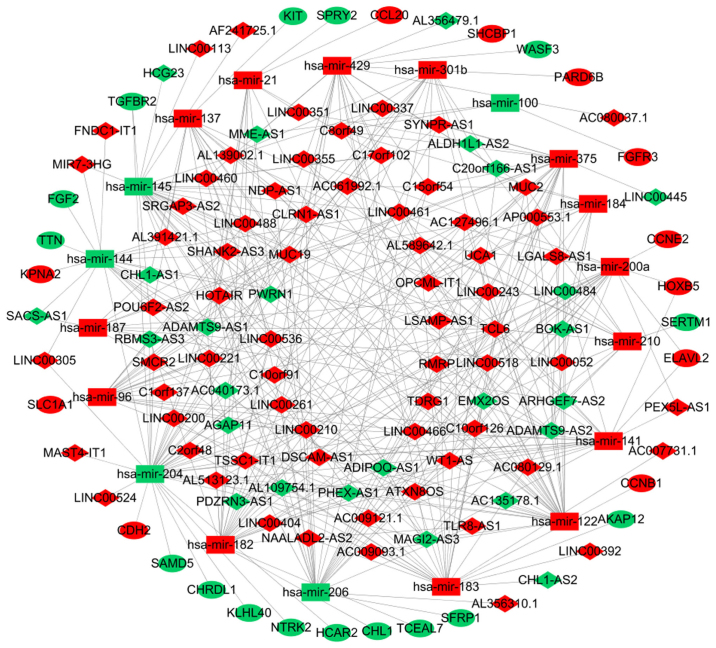
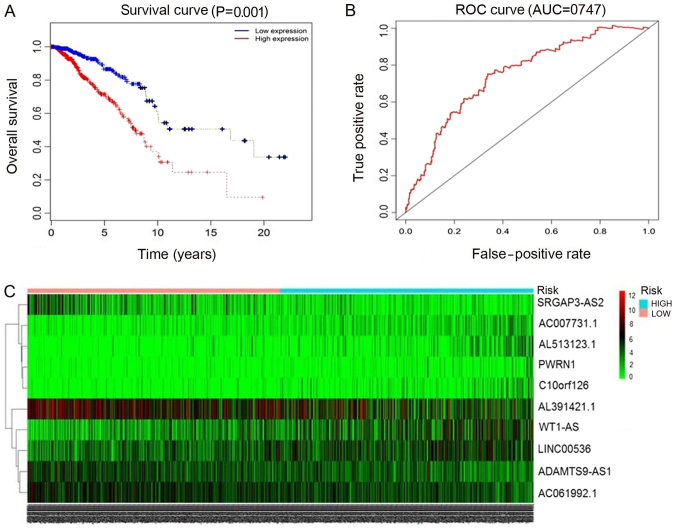
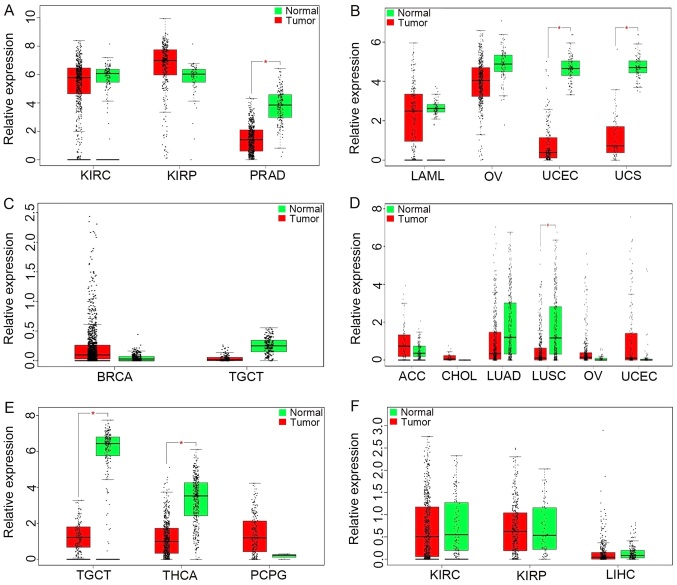
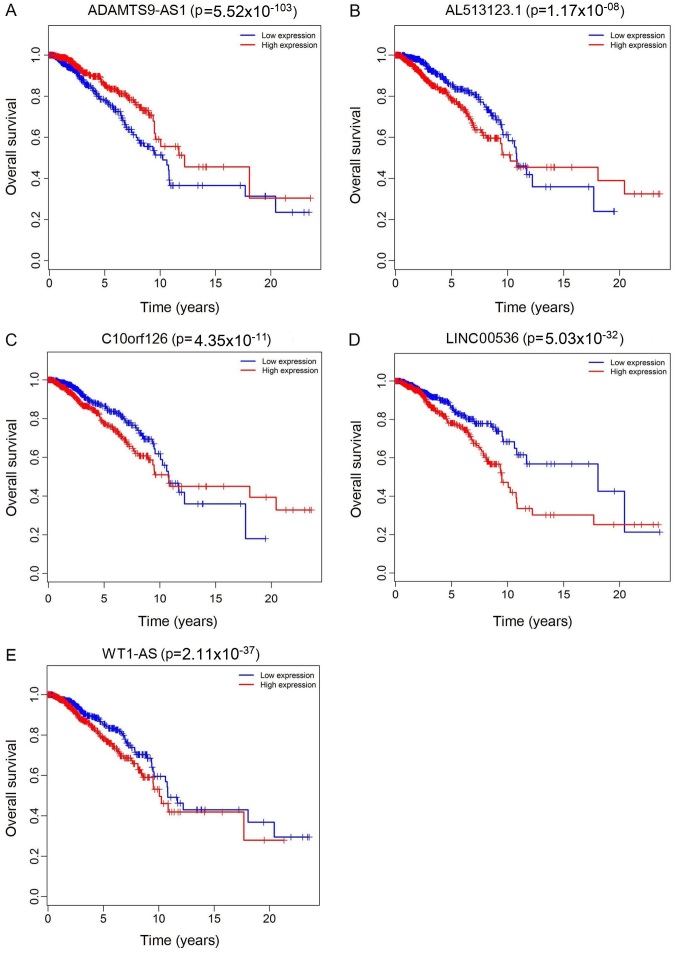
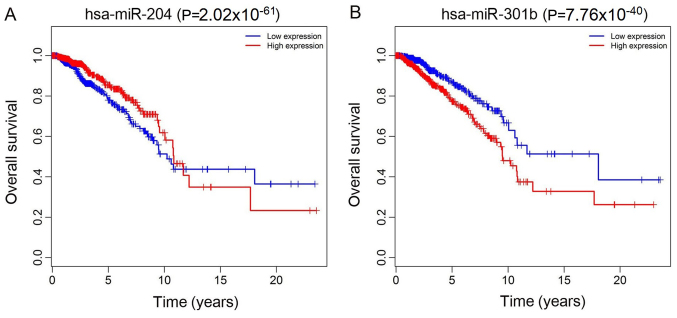
Previous studies have suggested that long non‑coding RNAs (lncRNAs) are closely associated with human diseases, particularly cancer, including cancer of the lung, breast and stomach. A variety of lncRNAs are abnormally expressed in cancer and participate in several pathways including cell proliferation and apoptosis; these elements are closely associated with the development of cancer. The Cancer Genome Atlas (TCGA) is an important cancer database. It consists of clinical data, genomic variation, mRNA, microRNA (miRNA) and lncRNAs expression, methylation and other data for various types of human cancer. In the present study, differential expression of RNA was identified using the edgeR package. A total 1,222 RNA sequencing profiles from patients with breast cancer were downloaded from TCGA. A competing endogenous RNA (ceRNA) network was constructed for breast cancer based on miRcode and miRTarBase. The top 10 lncRNAs were selected using Cox regression analysis. Survival analysis was performed using Kaplan‑Meier analysis. A total of 1,028 breast cancer‑associated lncRNAs and 89 miRNAs (fold change >2; P<0.05) were identified; among these, 93 lncRNAs and 19 miRNAs were included in the ceRNA network. Subsequently, 10 basic lncRNAs were selected and their associations with overall survival were identified. In addition, 5 lncRNAs (ADAM metallopeptidase with thrombospondin type 1 motif 9‑antisense RNA 1, AL513123.1, chromosome 10 open reading frame 126, long intergenic non‑protein coding RNA 536 and Wilms tumor 1 antisense RNA) were identified to be significantly associated with overall survival (P<0.05, log rank test). These results suggested that mRNAs, lncRNAs and miRNAs were involved in pathological mechanisms of breast cancer. The newly‑identified ceRNA network included 93 breast cancer‑specific lncRNAs, 19 miRNAs and 27 mRNAs. The results of the present study highlight the potential of lncRNAs in understanding the development and pathogenesis of breast cancer, and suggest novel concepts and an experimental basis for the identification of prognostic biomarkers and therapeutic targets for breast cancer.
先前的研究表明,长非编码 RNA(lncRNA)与人类疾病密切相关,尤其是癌症,包括肺癌、乳腺癌和胃癌。多种 lncRNA 在癌症中异常表达,并参与包括细胞增殖和凋亡在内的多种途径;这些因素与癌症的发展密切相关。癌症基因组图谱(TCGA)是一个重要的癌症数据库。它包含各种人类癌症的临床数据、基因组变异、mRNA、microRNA(miRNA)和 lncRNA 表达、甲基化等数据。在本研究中,使用 edgeR 软件包鉴定了 RNA 的差异表达。从 TCGA 下载了 1222 例乳腺癌患者的 RNA 测序谱。基于 miRcode 和 miRTarBase 构建了乳腺癌的竞争性内源 RNA(ceRNA)网络。使用 Cox 回归分析选择前 10 个 lncRNA。使用 Kaplan-Meier 分析进行生存分析。鉴定出 1028 个与乳腺癌相关的 lncRNA 和 89 个 miRNA(fold change >2;P<0.05);其中,93 个 lncRNA 和 19 个 miRNA 包含在 ceRNA 网络中。随后,选择了 10 个基础 lncRNA,并确定其与总生存的关系。此外,还鉴定了 5 个 lncRNA(ADAM 金属肽酶与血小板反应蛋白 1 型基序 9 反义 RNA 1、AL513123.1、染色体 10 开放阅读框 126、长非蛋白编码 RNA 536 和维尔姆斯瘤 1 反义 RNA)与总生存显著相关(P<0.05,对数秩检验)。这些结果表明,mRNA、lncRNA 和 miRNA 参与了乳腺癌的病理机制。新鉴定的 ceRNA 网络包含 93 个乳腺癌特异性 lncRNA、19 个 miRNA 和 27 个 mRNA。本研究的结果强调了 lncRNA 在理解乳腺癌发生和发病机制中的潜力,并为乳腺癌预后生物标志物和治疗靶点的识别提供了新的概念和实验依据。