West James D, Chen Xinping, Ping Ling, Gladson Santhi, Hamid Rizwan, Lloyd James E, Talati Megha
Vanderbilt University Medical Center.
Vanderbilt University School of Medicine.
Pulm Circ. 2019 May 24;10(1):2045894019856483. doi: 10.1177/2045894019856483.
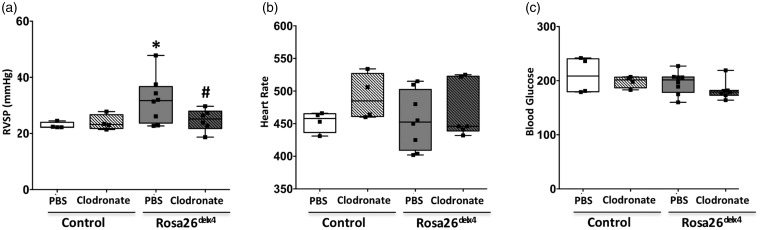
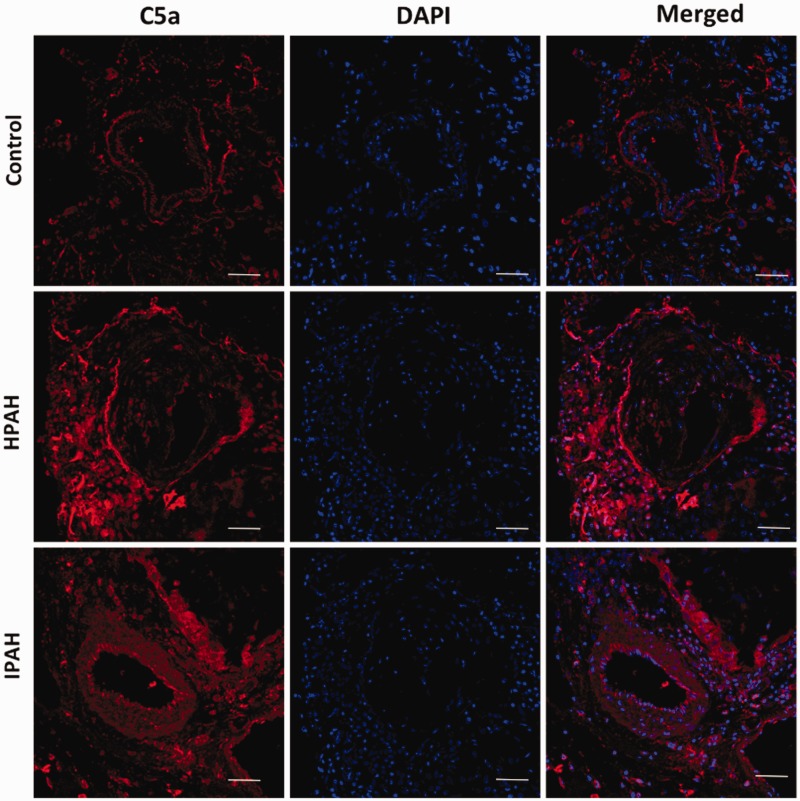
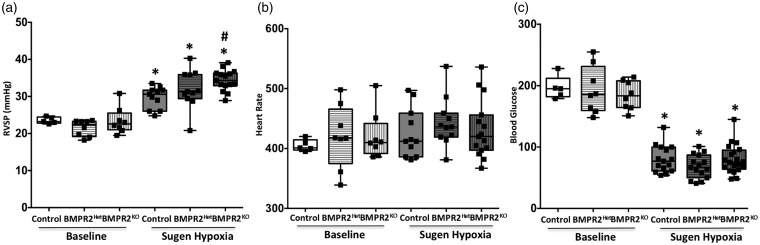
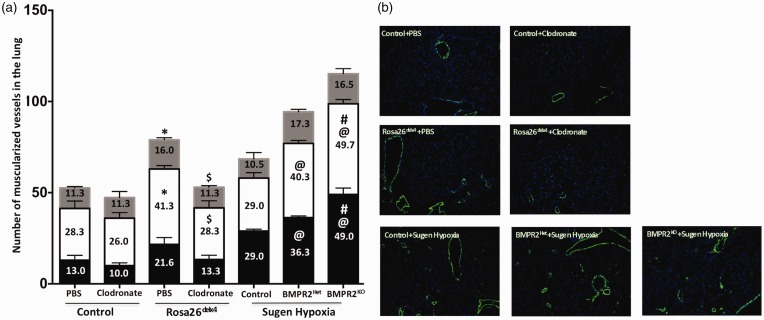
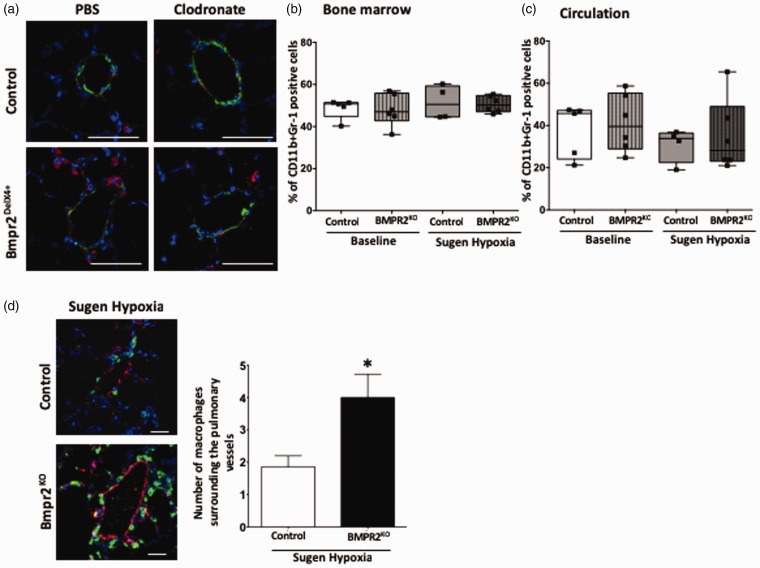
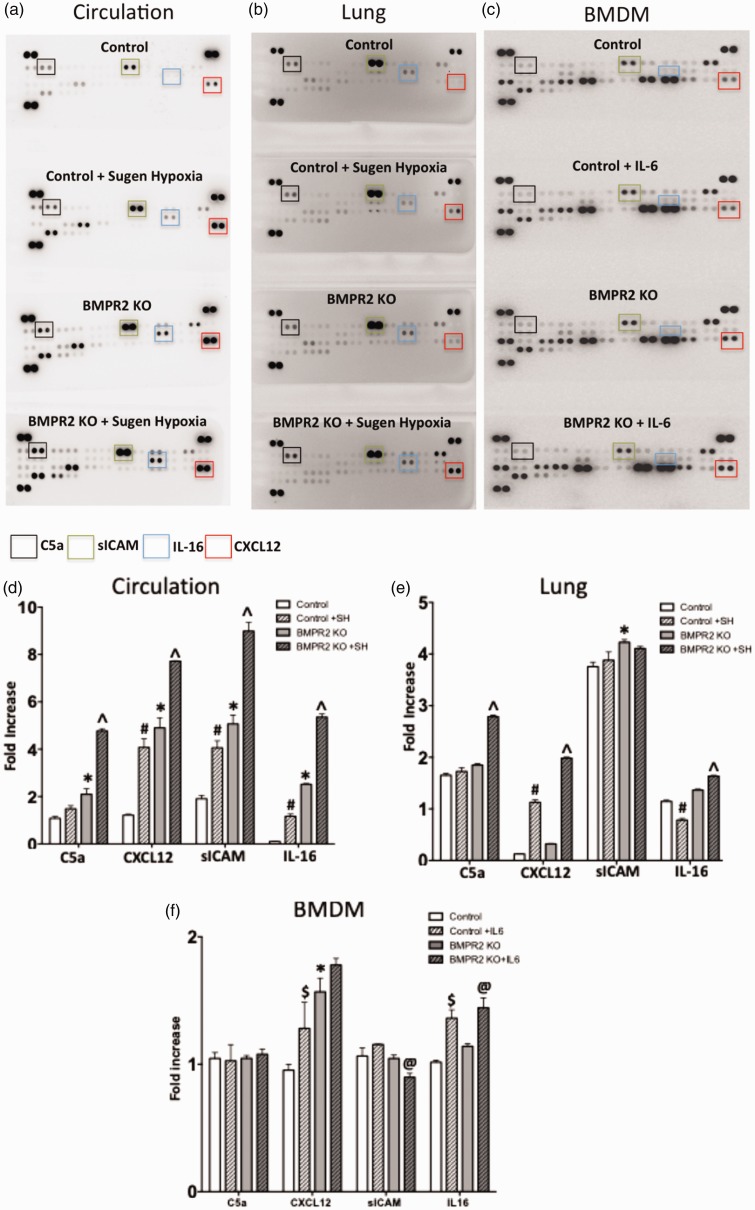
Inflammatory cells contribute to irreversible damage in pulmonary arterial hypertension (PAH). We hypothesized that in PAH, dysfunctional BMPR2 signaling in macrophages contributes to pulmonary vascular injury and phenotypic changes via proinflammatory cytokine production. Studies were conducted in: (1) Rosa26-rtTA2 3 X TetO7-Bmpr2delx4 FVB/N mice (mutant Bmpr2 is universally expressed, BMPR2 mice) given a weekly intra-tracheal liposomal clodronate injections for four weeks; and (2) LysM-Cre X floxed BMPR2 X floxed eGFP monocyte lineage-specific BMPR2 knockout (KO) mouse model (Bmpr2 gene expression knockdown in monocytic lineage cells) (BMPR2) following three weeks of sugen/hypoxia treatment. In the BMPR2 mice, increased right ventricular systolic pressure (RVSP; < 0.05) was normalized by clodronate, and in monocyte lineage-specific BMPR2 mice sugen hypoxia treatment increased ( < 0.05) RVSP compared to control littermates, suggesting that suppressed BMPR2 in macrophages modulate RVSP in animal models of PH. In addition, in these mouse models, muscularized pulmonary vessels were increased ( < 0.05) and surrounded by an increased number of macrophages. Elimination of macrophages in BMPR2 mice reduced the number of muscularized pulmonary vessels and macrophages surrounding these vessels. Further, in monocyte lineage-specific BMPR2 mice, there was significant increase in proinflammatory cytokines, including C-X-C Motif Chemokine Ligand 12 (CXCL12), complement component 5 a (C5a), Interleukin-16 (IL-16), and secretory ICAM. C5a positive inflammatory cells present in and around the pulmonary vessels in the PAH lung could potentially be involved in pulmonary vessel remodeling. In summary, our data indicate that, in BMPR2-related PAH, macrophages with dysfunctional BMPR2 influence pulmonary vascular remodeling and phenotypic outcomes via proinflammatory cytokine production.
炎症细胞在肺动脉高压(PAH)中导致不可逆损伤。我们假设,在PAH中,巨噬细胞中功能失调的骨形态发生蛋白受体2(BMPR2)信号传导通过促炎细胞因子的产生导致肺血管损伤和表型变化。研究在以下模型中进行:(1)Rosa26-rtTA2 3 X TetO7-Bmpr2delx4 FVB/N小鼠(突变型Bmpr2普遍表达,即BMPR2小鼠),每周经气管内注射脂质体氯膦酸盐,持续四周;(2)LysM-Cre X floxed BMPR2 X floxed eGFP单核细胞谱系特异性BMPR2基因敲除(KO)小鼠模型(单核细胞谱系细胞中Bmpr2基因表达下调)(BMPR2),经苏氨酸/缺氧处理三周。在BMPR2小鼠中,氯膦酸盐使升高的右心室收缩压(RVSP;<0.05)恢复正常,与对照同窝小鼠相比,单核细胞谱系特异性BMPR2小鼠经苏氨酸缺氧处理后RVSP升高(<0.05),这表明巨噬细胞中BMPR2的抑制调节了PH动物模型中的RVSP。此外,在这些小鼠模型中,肺血管肌化增加(<0.05),周围巨噬细胞数量增加。消除BMPR2小鼠中的巨噬细胞减少了肺血管肌化的数量以及围绕这些血管的巨噬细胞数量。此外,在单核细胞谱系特异性BMPR2小鼠中,促炎细胞因子显著增加,包括C-X-C基序趋化因子配体12(CXCL12)、补体成分5a(C5a)、白细胞介素-16(IL-16)和分泌型细胞间黏附分子。PAH肺中肺血管及其周围存在的C5a阳性炎症细胞可能参与肺血管重塑。总之,我们的数据表明,在与BMPR2相关的PAH中,BMPR2功能失调的巨噬细胞通过促炎细胞因子的产生影响肺血管重塑和表型结果。