Graduate Institute of Cancer Molecular Biology and Drug Discovery, College of Medical Science and Technology, Taipei Medical University, Taipei, Taiwan.
Pharmacological Institute, College of Medicine, National Taiwan University, Taipei, Taiwan.
Clin Epigenetics. 2019 May 29;11(1):85. doi: 10.1186/s13148-019-0681-6.
Oncogenic K-Ras signaling highly relies on the canonical Ras/MEK/ERK pathway to contribute to pancreatic cancer progression. However, numerous efforts of MEK inhibitors have failed to provide an optimal antitumor effect for pancreatic cancer in practice. The aim of the present work was to develop a more efficacious therapeutic intervention for MEK inhibitors through combination with histone deacetylase (HDAC) inhibitor MPT0E028.
The effects of combined therapy on cell viability, apoptosis, protein, and RNA expressions were determined by MTT assay, flow cytometry, western blotting, and quantitative PCR analysis. The AsPC-1 xenograft was used to assess antitumor effects in vivo.
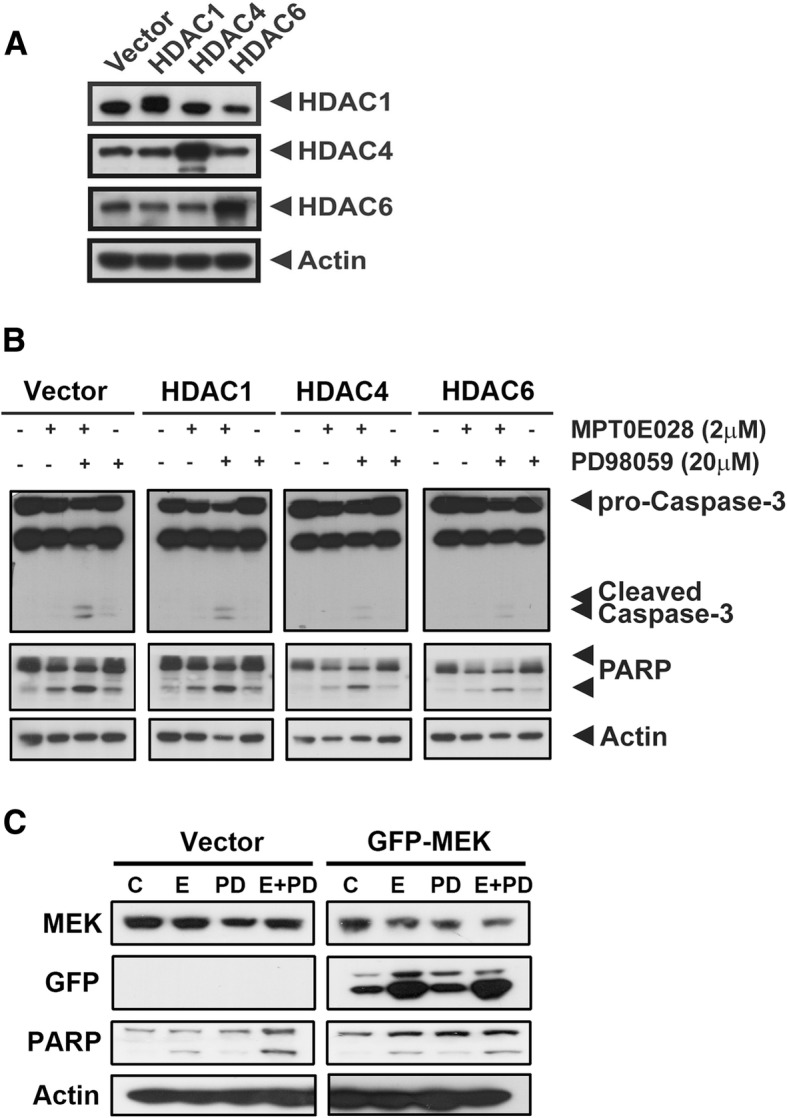
The co-administration of MPT0E028 and MEK inhibitor yielded synergistic effects on cell viability suppression both in K-Ras mutated and wild-type pancreatic cancer cells and also markedly triggered cell apoptosis. Surprisingly, ERK and epidermal growth factor receptor (EGFR) were activated by the long-term and low-concentration treatment of MPT0E028 or another HDAC inhibitor alone. Whereas, the pharmacological attenuation of ERK signaling dramatically abolished the MPTE028-induced p-ERK and EGFR expression. Overexpression of HDAC4, HDAC6, and MEK, respectively, reversed the cell death induced by the combined treatment. Finally, the combined treatment decreased the tumor volume in an AsPC-1 xenograft model compared to each individual treatment alone.
The synergistic anti-survival effect of the combination was suggested to occur via compensation of the MEK inhibitor for activated ERK. Our results indicate that this combination strategy could benefit patients with pancreatic cancer beyond K-Ras status.
致癌性 K-Ras 信号高度依赖经典的 Ras/MEK/ERK 通路,有助于胰腺癌的进展。然而,MEK 抑制剂的众多努力在实践中未能为胰腺癌提供最佳的抗肿瘤效果。本研究旨在通过与组蛋白去乙酰化酶(HDAC)抑制剂 MPT0E028 联合开发更有效的 MEK 抑制剂治疗干预措施。
通过 MTT 测定法、流式细胞术、Western blot 和定量 PCR 分析确定联合治疗对细胞活力、凋亡、蛋白质和 RNA 表达的影响。使用 AsPC-1 异种移植模型评估体内抗肿瘤作用。
MPT0E028 和 MEK 抑制剂联合给药对 K-Ras 突变和野生型胰腺癌细胞的细胞活力抑制具有协同作用,并且明显触发细胞凋亡。令人惊讶的是,ERK 和表皮生长因子受体(EGFR)被 MPT0E028 或另一种 HDAC 抑制剂的长期和低浓度处理激活。然而,ERK 信号的药理学抑制显著消除了 MPTE028 诱导的 p-ERK 和 EGFR 表达。分别过表达 HDAC4、HDAC6 和 MEK 可逆转联合处理诱导的细胞死亡。最后,与单独治疗相比,联合治疗可降低 AsPC-1 异种移植模型中的肿瘤体积。
联合治疗的协同抗生存作用提示通过 MEK 抑制剂补偿激活的 ERK 发生。我们的结果表明,这种联合策略可能使无论 K-Ras 状态如何的胰腺癌患者受益。