Department of Clinical Genetics, Children's Hospital at Westmead, Sydney Children's Hospital Network, Sydney, New South Wales, Australia.
Discipline of Genomic Medicine, Sydney Medical School, University of Sydney, Sydney, New South Wales, Australia.
Am J Med Genet A. 2019 Aug;179(8):1585-1590. doi: 10.1002/ajmg.a.61200. Epub 2019 Jun 7.
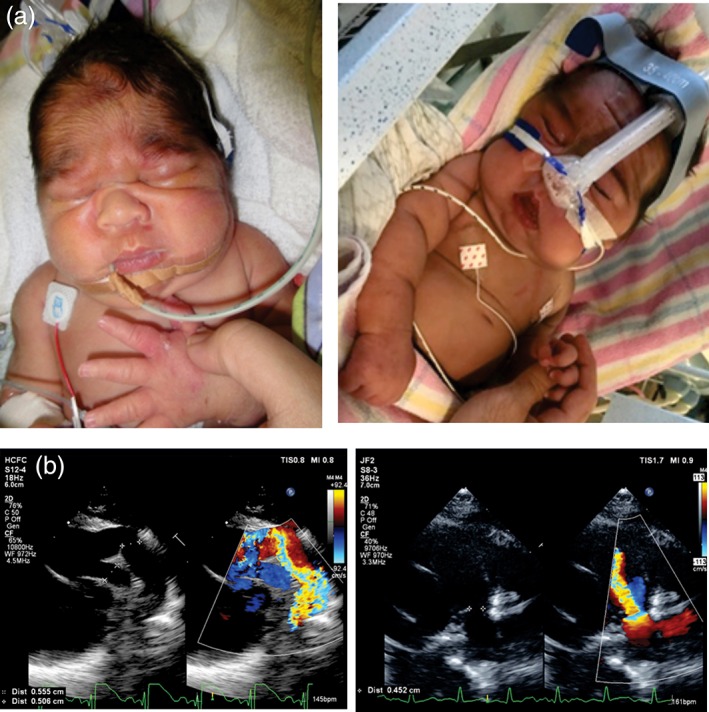
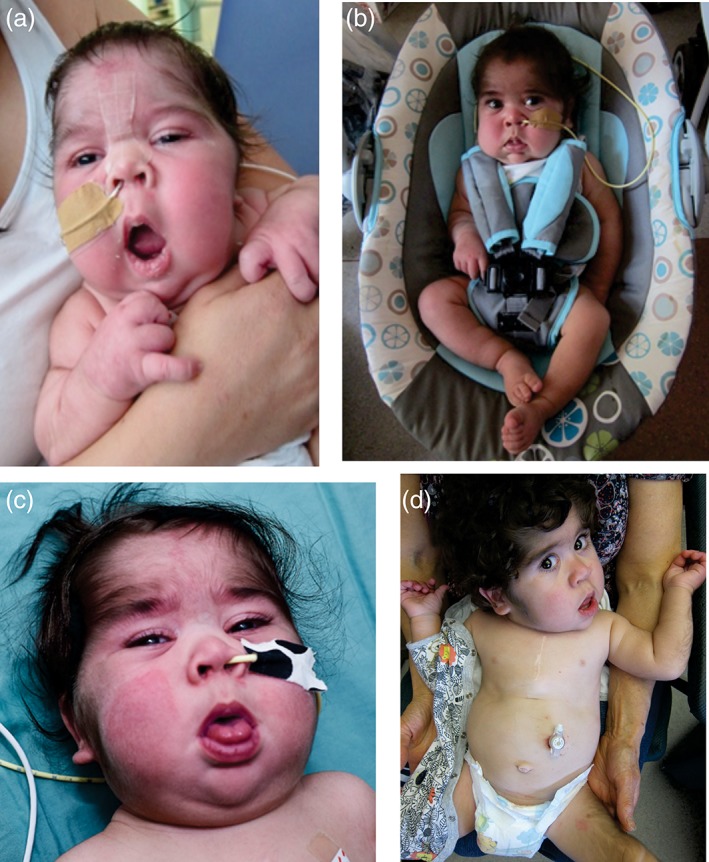
Cantú syndrome (CS), characterized by hypertrichosis, distinctive facial features, and complex cardiovascular abnormalities, is caused by pathogenic variants in ABCC9 and KCNJ8 genes. These genes encode gain-of-function mutations in the regulatory (SUR2) and pore-forming (Kir6.1) subunits of K channels, respectively, suggesting that channel-blocking sulfonylureas could be a viable therapy. Here we report a neonate with CS, carrying a heterozygous ABCC9 variant (c.3347G>A, p.Arg1116His), born prematurely at 32 weeks gestation. Initial echocardiogram revealed a large patent ductus arteriosus (PDA), and high pulmonary pressures with enlarged right ventricle. He initially received surfactant and continuous positive airway pressure ventilation and was invasively ventilated for 4 weeks, until PDA ligation. After surgery, he still had ongoing bilevel positive airway pressure (BiPAP) requirement, but was subsequently weaned to nocturnal BiPAP. He was treated for pulmonary hypertension with Sildenafil, but failed to make further clinical improvement. A therapeutic glibenclamide trial was commenced in week 11 (initial dose of 0.05 mg kg day in two divided doses). After 1 week of treatment, he began to tolerate time off BiPAP when awake, and edema improved. Glibenclamide was well tolerated, and the dose was slowly increased to 0.15 mg kg day over the next 12 weeks. Mild transient hypoglycemia was observed, but there was no cardiovascular dysfunction. Confirmation of therapeutic benefit will require studies of more CS patients but, based on this limited experience, consideration should be given to glibenclamide as CS therapy, although problems associated with prematurity, and complications of hypoglycemia, might limit outcome in critically ill neonates with CS.
坎图综合征(CS)的特征是多毛症、独特的面部特征和复杂的心血管异常,由 ABCC9 和 KCNJ8 基因的致病性变异引起。这些基因分别编码 K 通道的调节(SUR2)和孔形成(Kir6.1)亚基的功能获得性突变,这表明通道阻断磺酰脲类药物可能是一种可行的治疗方法。在这里,我们报告了一例 CS 新生儿,携带 ABCC9 杂合变异(c.3347G>A,p.Arg1116His),妊娠 32 周早产。最初的超声心动图显示大动脉导管未闭(PDA)较大,肺动脉压高,右心室增大。他最初接受了表面活性剂和持续气道正压通气,并进行了 4 周的有创通气,直到 PDA 结扎。手术后,他仍需要持续双水平气道正压通气(BiPAP),但随后逐渐减少为夜间 BiPAP。他接受了西地那非治疗肺动脉高压,但没有进一步的临床改善。在第 11 周开始进行治疗性格列本脲试验(初始剂量为 0.05mg/kg/天,分两次服用)。治疗 1 周后,他开始在清醒时耐受 BiPAP 停机,水肿改善。格列本脲耐受良好,剂量在接下来的 12 周内缓慢增加至 0.15mg/kg/天。观察到轻微的短暂性低血糖,但没有心血管功能障碍。更多 CS 患者的研究将需要确认治疗效果,但基于这一有限的经验,应考虑将格列本脲作为 CS 的治疗方法,尽管与早产儿相关的问题和低血糖并发症可能会限制患有 CS 的危重新生儿的预后。