Zepeda-Mendoza Cinthya, Goodenberger McKinsey L, Kuhl Ashley, Rice Gregory M, Hoppman Nicole
Division of Laboratory Genetics and Genomics, Departments of Laboratory Medicine and Pathology Mayo Clinic Rochester Minnesota.
School of Medicine and Public Health University of Wisconsin Madison Wisconsin.
Clin Case Rep. 2019 May 4;7(6):1154-1160. doi: 10.1002/ccr3.2186. eCollection 2019 Jun.
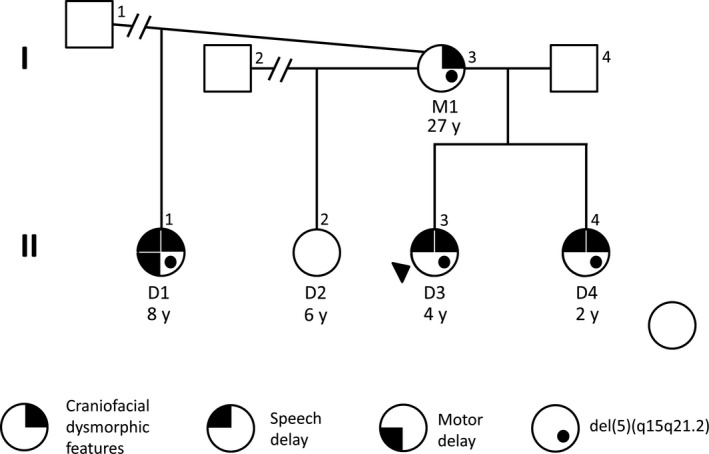
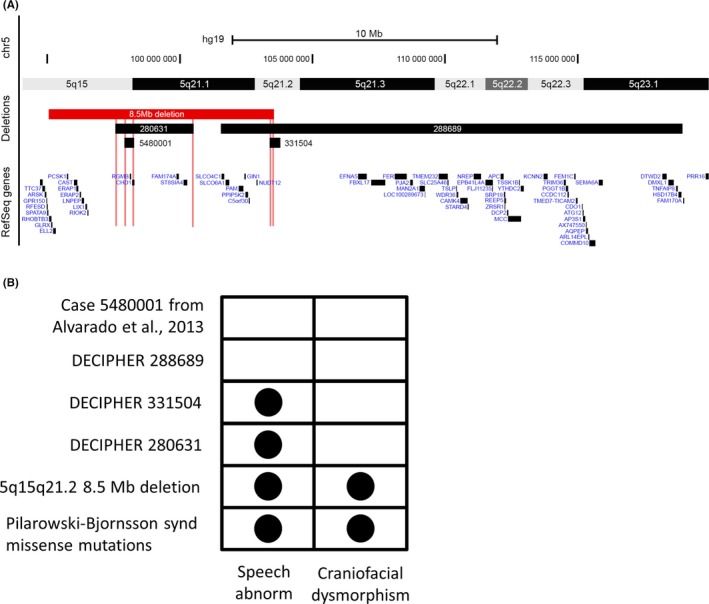
We report a two-generation family with four females harboring an 8.5Mb heterozygous deletion of 5q15-q21.2 who present with dysmorphic craniofacial features and speech delay. We hypothesize haploinsufficiency of to be contributing to the clinical features observed in this family.
我们报告了一个两代家族,其中四名女性携带5q15 - q21.2区域8.5Mb的杂合性缺失,她们表现出颅面部畸形特征和语言发育迟缓。我们推测该区域的单倍剂量不足是导致这个家族中观察到的临床特征的原因。