Population Health and Immunity Division, The Walter and Eliza Hall Institute of Medical Research, 1G Royal Parade, Parkville, VIC 3052, Australia; Department of Medical Biology, University of Melbourne, 1G Royal Parade, Parkville, VIC 3052, Australia.
Cerebellar Ataxia Clinic, Neuroscience Department, Alfred Health, Melbourne, VIC 3004, Australia; Balance Disorders and Ataxia Service, Royal Victorian Eye & Ear Hospital, East Melbourne, VIC 3002, Australia.
Am J Hum Genet. 2019 Jul 3;105(1):151-165. doi: 10.1016/j.ajhg.2019.05.016. Epub 2019 Jun 20.
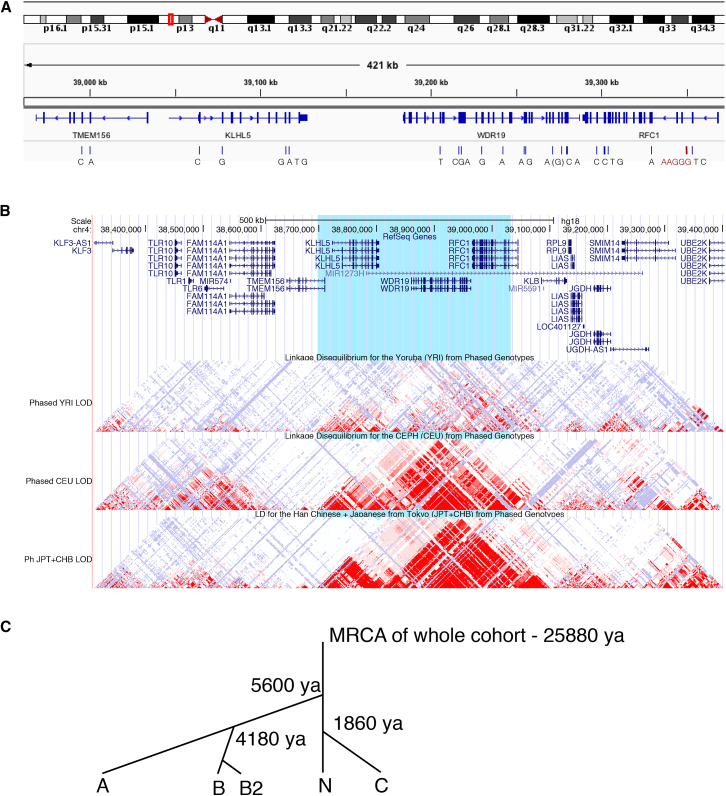
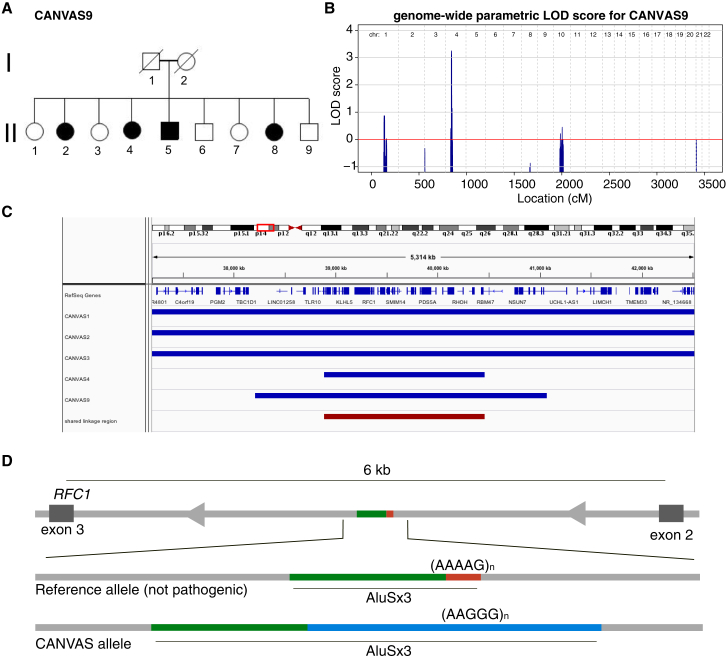
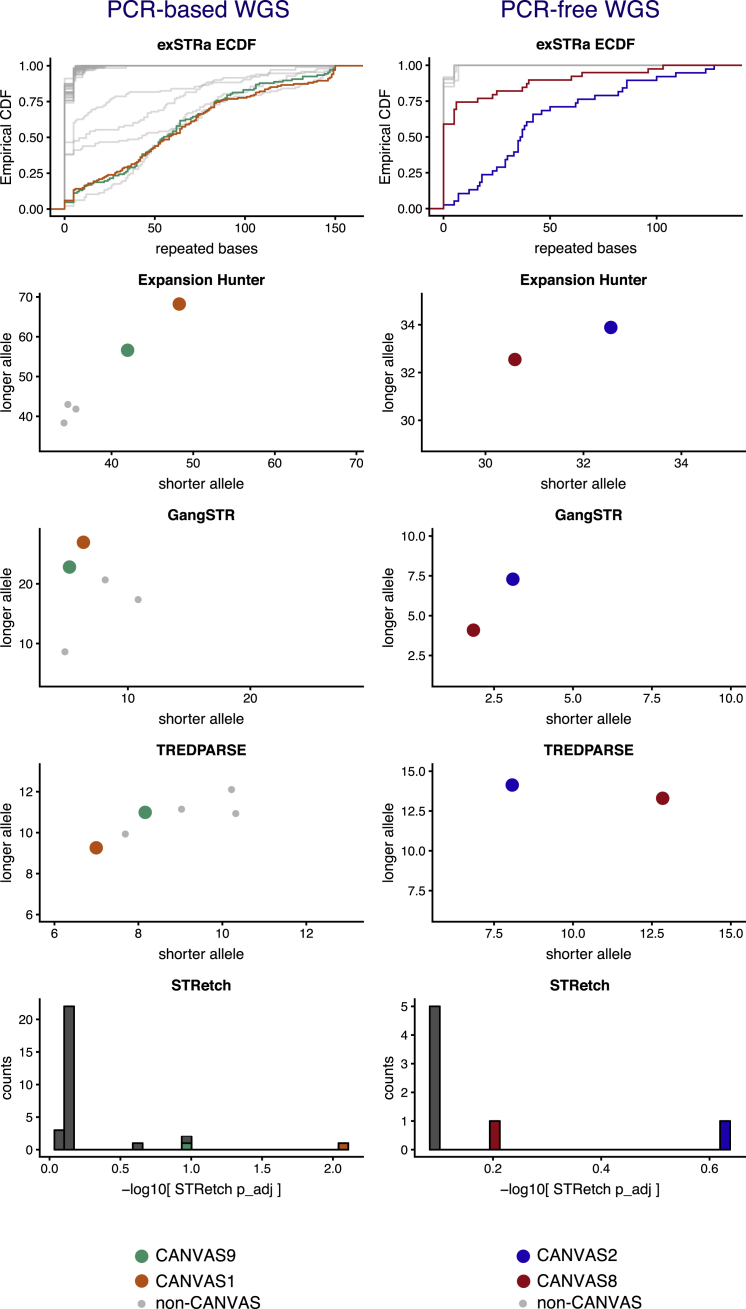
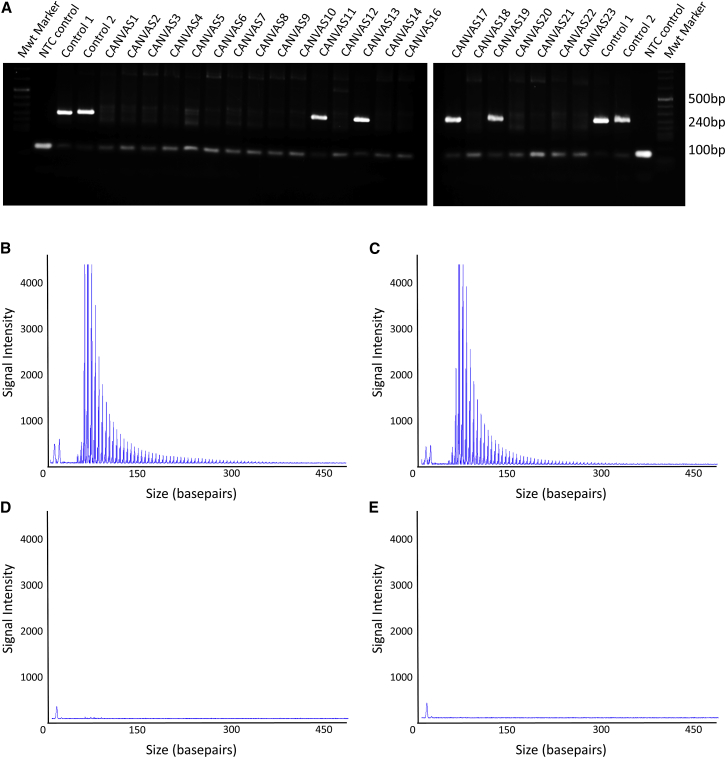
Genomic technologies such as next-generation sequencing (NGS) are revolutionizing molecular diagnostics and clinical medicine. However, these approaches have proven inefficient at identifying pathogenic repeat expansions. Here, we apply a collection of bioinformatics tools that can be utilized to identify either known or novel expanded repeat sequences in NGS data. We performed genetic studies of a cohort of 35 individuals from 22 families with a clinical diagnosis of cerebellar ataxia with neuropathy and bilateral vestibular areflexia syndrome (CANVAS). Analysis of whole-genome sequence (WGS) data with five independent algorithms identified a recessively inherited intronic repeat expansion [(AAGGG)] in the gene encoding Replication Factor C1 (RFC1). This motif, not reported in the reference sequence, localized to an Alu element and replaced the reference (AAAAG) short tandem repeat. Genetic analyses confirmed the pathogenic expansion in 18 of 22 CANVAS-affected families and identified a core ancestral haplotype, estimated to have arisen in Europe more than twenty-five thousand years ago. WGS of the four RFC1-negative CANVAS-affected families identified plausible variants in three, with genomic re-diagnosis of SCA3, spastic ataxia of the Charlevoix-Saguenay type, and SCA45. This study identified the genetic basis of CANVAS and demonstrated that these improved bioinformatics tools increase the diagnostic utility of WGS to determine the genetic basis of a heterogeneous group of clinically overlapping neurogenetic disorders.
基因组技术,如下一代测序(NGS),正在彻底改变分子诊断学和临床医学。然而,这些方法在识别致病性重复扩展方面已经被证明效率低下。在这里,我们应用了一系列生物信息学工具,可以用于在 NGS 数据中识别已知或新的扩展重复序列。我们对 35 名来自 22 个家族的个体进行了遗传研究,这些个体的临床诊断为小脑共济失调伴神经病和双侧前庭反射消失综合征(CANVAS)。对全基因组序列(WGS)数据的分析使用了五种独立的算法,鉴定出编码复制因子 C1(RFC1)的基因中的隐性遗传内含子重复扩展 [(AAGGG)]。该基序未在参考序列中报道,定位于 Alu 元件并取代了参考(AAAAG)短串联重复序列。遗传分析在 22 个 CANVAS 受影响的家族中的 18 个中证实了致病性扩展,并鉴定了一个核心祖先单倍型,估计它在 25000 多年前出现在欧洲。对四个 RFC1 阴性的 CANVAS 受影响的家族的 WGS 进行了分析,在三个家族中发现了合理的变体,对 SCA3、Charlevoix-Saguenay 型痉挛性共济失调和 SCA45 进行了基因组重新诊断。本研究确定了 CANVAS 的遗传基础,并证明了这些改进的生物信息学工具增加了 WGS 的诊断效用,以确定一组临床重叠的神经遗传疾病的遗传基础。