Lai Hong-Yan, Zhang Zhao-Yue, Su Zhen-Dong, Su Wei, Ding Hui, Chen Wei, Lin Hao
Key Laboratory for NeuroInformation of Ministry of Education, School of Life Science and Technology and Center for Informational Biology, University of Electronic Science and Technology of China, Chengdu 610054, China.
Key Laboratory for NeuroInformation of Ministry of Education, School of Life Science and Technology and Center for Informational Biology, University of Electronic Science and Technology of China, Chengdu 610054, China; Innovative Institute of Chinese Medicine and Pharmacy, Chengdu University of Traditional Chinese Medicine, Chengdu 611730, China; Center for Genomics and Computational Biology, School of Life Sciences, North China University of Science and Technology, Tangshan 063000, China.
Mol Ther Nucleic Acids. 2019 Sep 6;17:337-346. doi: 10.1016/j.omtn.2019.05.028. Epub 2019 Jun 13.
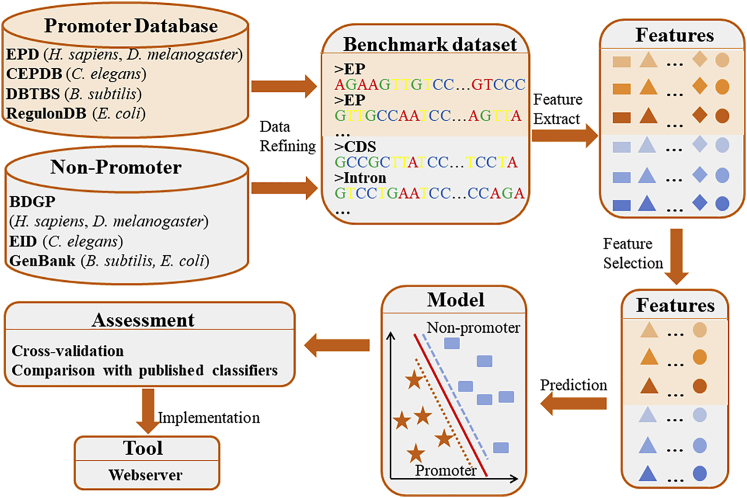
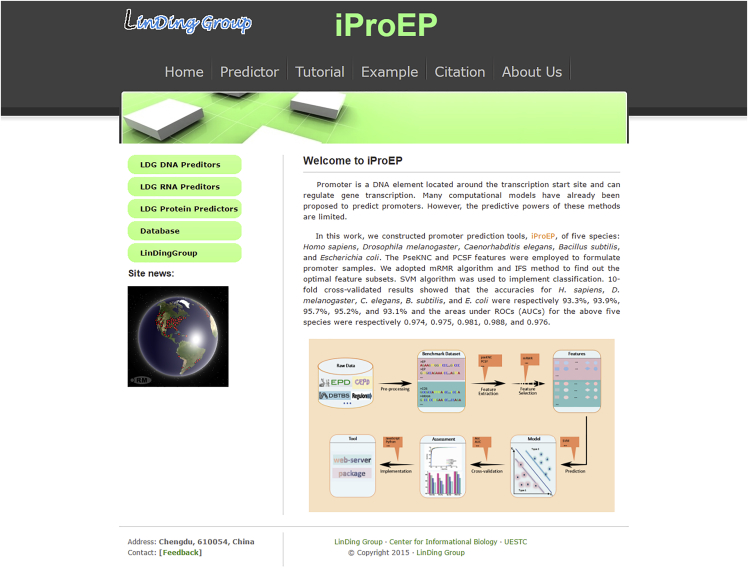
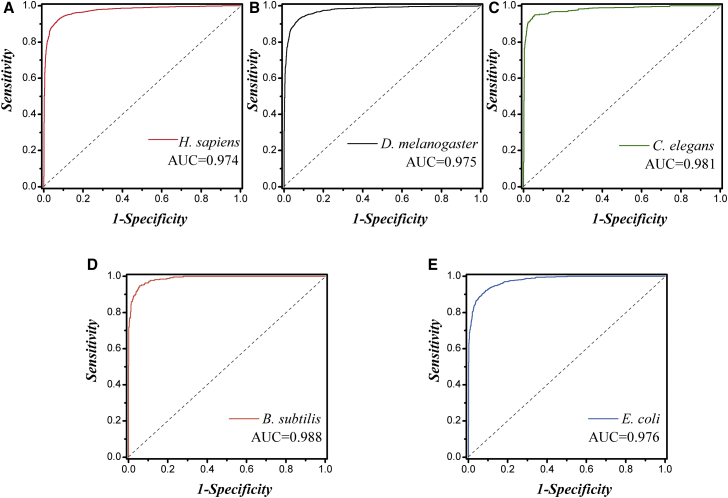
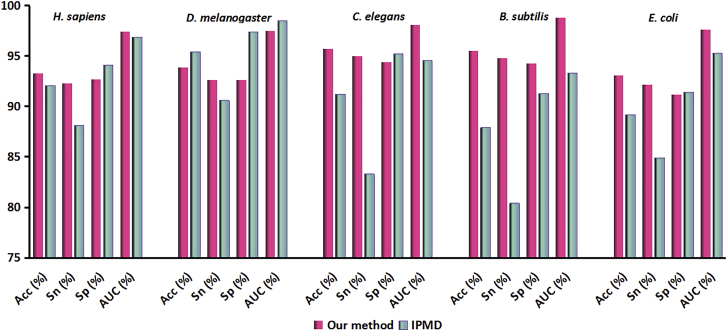
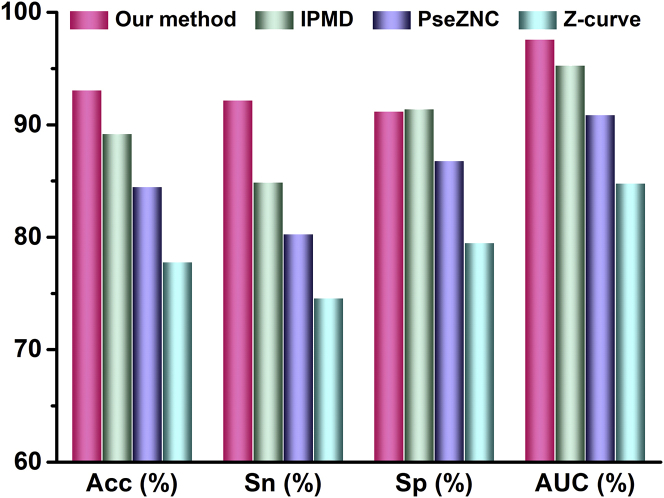
Promoter is a fundamental DNA element located around the transcription start site (TSS) and could regulate gene transcription. Promoter recognition is of great significance in determining transcription units, studying gene structure, analyzing gene regulation mechanisms, and annotating gene functional information. Many models have already been proposed to predict promoters. However, the performances of these methods still need to be improved. In this work, we combined pseudo k-tuple nucleotide composition (PseKNC) with position-correlation scoring function (PCSF) to formulate promoter sequences of Homo sapiens (H. sapiens), Drosophila melanogaster (D. melanogaster), Caenorhabditis elegans (C. elegans), Bacillus subtilis (B. subtilis), and Escherichia coli (E. coli). Minimum Redundancy Maximum Relevance (mRMR) algorithm and increment feature selection strategy were then adopted to find out optimal feature subsets. Support vector machine (SVM) was used to distinguish between promoters and non-promoters. In the 10-fold cross-validation test, accuracies of 93.3%, 93.9%, 95.7%, 95.2%, and 93.1% were obtained for H. sapiens, D. melanogaster, C. elegans, B. subtilis, and E. coli, with the areas under receiver operating curves (AUCs) of 0.974, 0.975, 0.981, 0.988, and 0.976, respectively. Comparative results demonstrated that our method outperforms existing methods for identifying promoters. An online web server was established that can be freely accessed (http://lin-group.cn/server/iProEP/).
启动子是位于转录起始位点(TSS)周围的一种基本DNA元件,能够调控基因转录。启动子识别在确定转录单元、研究基因结构、分析基因调控机制以及注释基因功能信息方面具有重要意义。已经提出了许多模型来预测启动子。然而,这些方法的性能仍有待提高。在这项工作中,我们将伪k元核苷酸组成(PseKNC)与位置相关评分函数(PCSF)相结合,构建了人类(智人)、黑腹果蝇、秀丽隐杆线虫、枯草芽孢杆菌和大肠杆菌的启动子序列。然后采用最小冗余最大相关(mRMR)算法和增量特征选择策略来找出最优特征子集。使用支持向量机(SVM)区分启动子和非启动子。在10折交叉验证测试中,智人、黑腹果蝇、秀丽隐杆线虫、枯草芽孢杆菌和大肠杆菌的准确率分别为93.3%、93.9%、95.7%、95.2%和93.1%,相应的受试者工作特征曲线下面积(AUC)分别为0.974、0.975、0.981、0.988和0.976。比较结果表明,我们的方法在识别启动子方面优于现有方法。我们建立了一个可免费访问的在线网络服务器(http://lin-group.cn/server/iProEP/)。