State Key Laboratory of Drug Research, The National Center for Drug Screening, CAS Key Laboratory of Receptor Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, 201203, China.
University of Chinese Academy of Sciences, Beijing, 100049, China.
Acta Pharmacol Sin. 2020 Mar;41(3):293-302. doi: 10.1038/s41401-019-0267-z. Epub 2019 Jul 17.
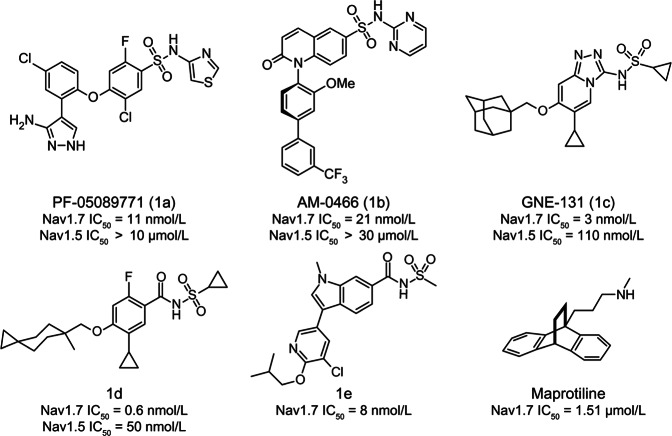
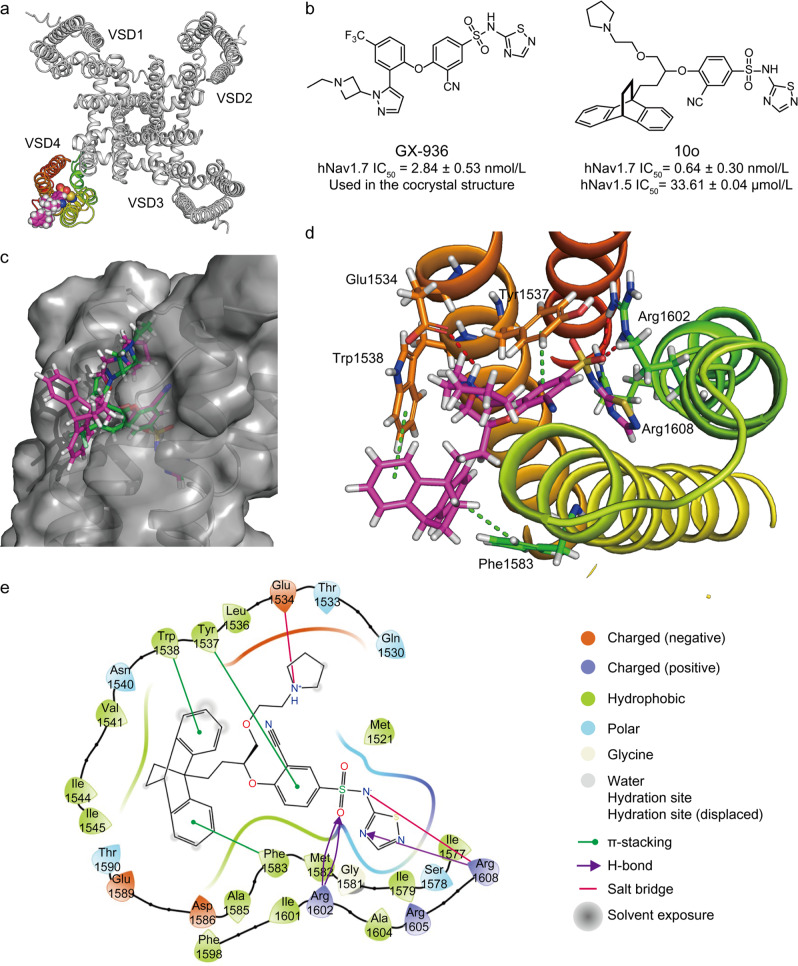
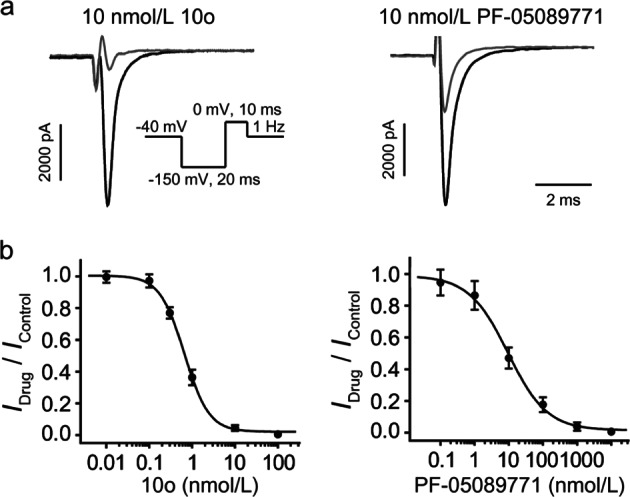
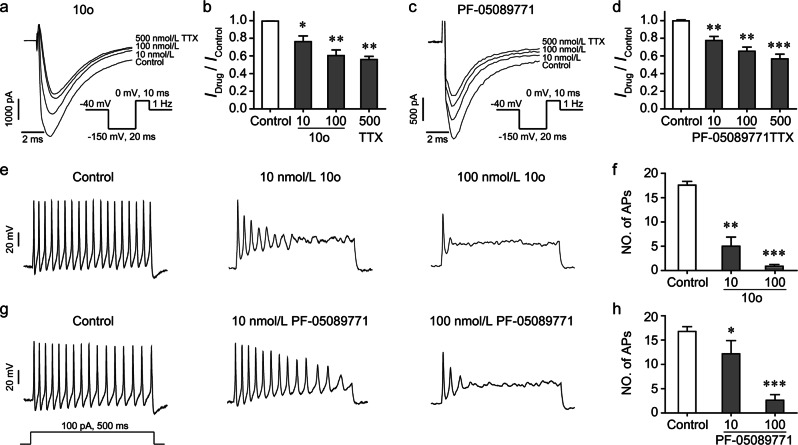
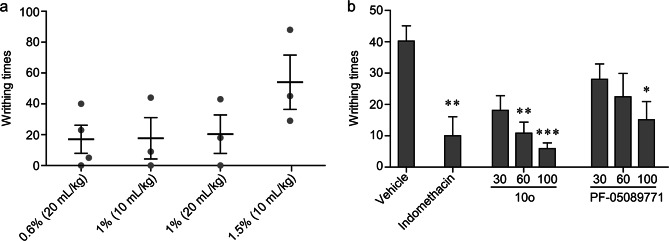
Nav1.7 channels are mainly distributed in the peripheral nervous system. Blockade of Nav1.7 channels with small-molecule inhibitors in humans might provide pain relief without affecting the central nervous system. Based on the facts that many reported Nav1.7-selective inhibitors contain aryl sulfonamide fragments, as well as a tricyclic antidepressant, maprotiline, has been found to inhibit Nav1.7 channels, we designed and synthesized a series of compounds with ethanoanthracene and aryl sulfonamide moieties. Their inhibitory activity on sodium channels were detected with electrophysiological techniques. We found that compound 10o potently inhibited Nav1.7 channels stably expressed in HEK293 cells (IC = 0.64 ± 0.30 nmol/L) and displayed a high Nav1.7/Nav1.5 selectivity. In mouse small-sized dorsal root ganglion neurons, compound 10o (10, 100 nmol/L) dose-dependently decreased the sodium currents and dramatically suppressed depolarizing current-elicited neuronal discharge. Preliminary in vivo experiments showed that compound 10o possessed good analgesic activity: in a mouse visceral pain model, administration of compound 10o (30-100 mg/kg, i.p.) effectively and dose-dependently suppressed acetic acid-induced writhing.
Nav1.7 通道主要分布在外周神经系统。用小分子抑制剂阻断 Nav1.7 通道可能会减轻疼痛而不影响中枢神经系统。基于许多报道的 Nav1.7 选择性抑制剂都含有芳基磺酰胺片段,以及三环抗抑郁药马普替林已被发现抑制 Nav1.7 通道的事实,我们设计并合成了一系列含有乙撑蒽和芳基磺酰胺部分的化合物。它们对钠离子通道的抑制活性通过电生理技术进行了检测。我们发现化合物 10o 能有效地抑制稳定表达在 HEK293 细胞中的 Nav1.7 通道(IC = 0.64 ± 0.30 nmol/L),并且表现出高的 Nav1.7/Nav1.5 选择性。在小鼠小尺寸背根神经节神经元中,化合物 10o(10、100 nmol/L)剂量依赖性地减少钠离子电流,并显著抑制去极化电流引起的神经元放电。初步的体内实验表明,化合物 10o 具有良好的镇痛活性:在小鼠内脏疼痛模型中,给予化合物 10o(30-100 mg/kg,腹腔注射)能有效和剂量依赖性地抑制醋酸引起的扭体反应。