Institute of Nephrology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, 450052, China; Division of Kidney Disease and Hypertension, Brown University School of Medicine, Providence, RI, 02903, United States; Division of Nephrology, University of Toledo College of Medicine, Toledo, OH, 43614, United States.
Division of Kidney Disease and Hypertension, Brown University School of Medicine, Providence, RI, 02903, United States.
Redox Biol. 2019 Sep;26:101275. doi: 10.1016/j.redox.2019.101275. Epub 2019 Jul 17.
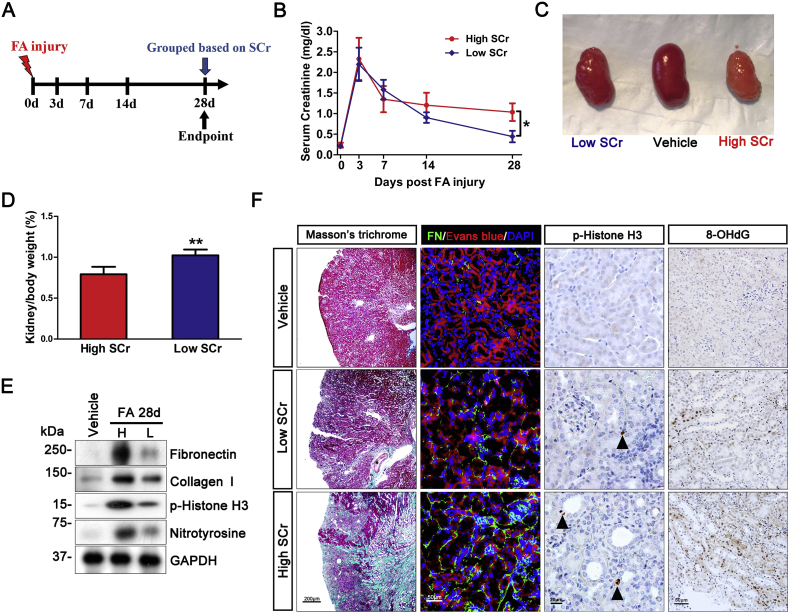
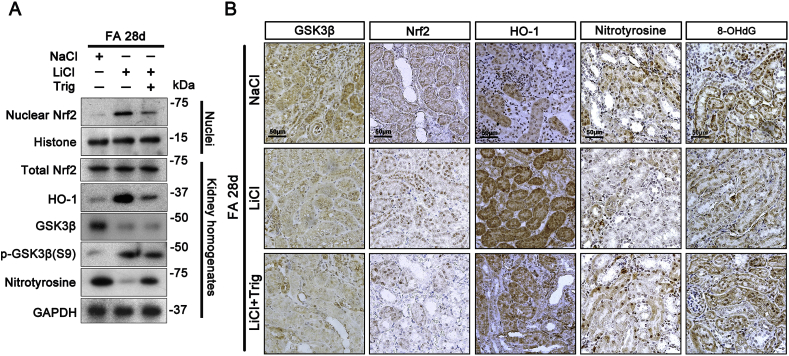
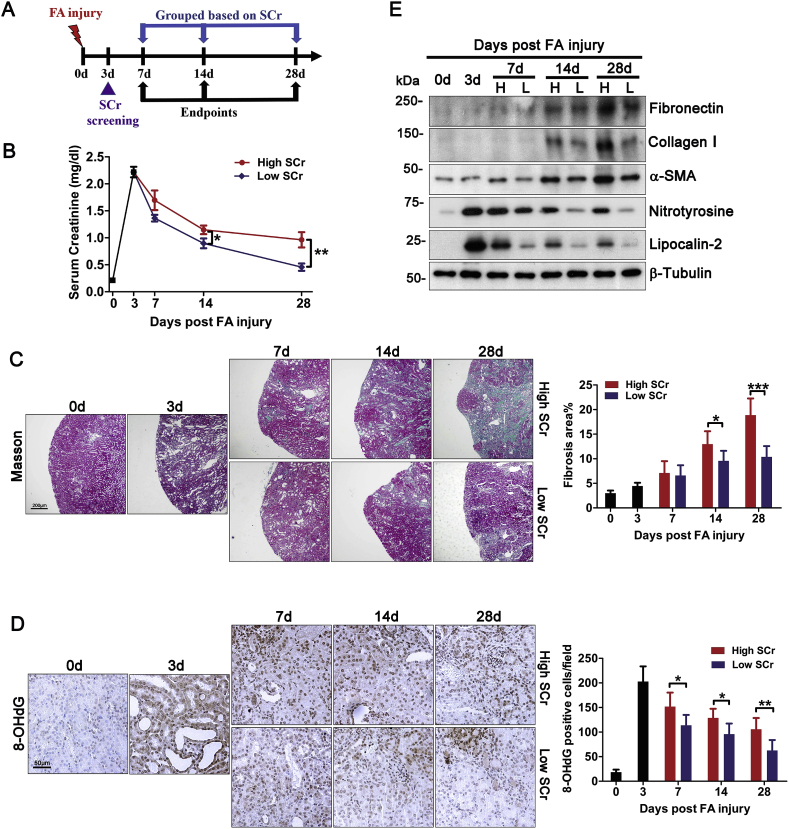
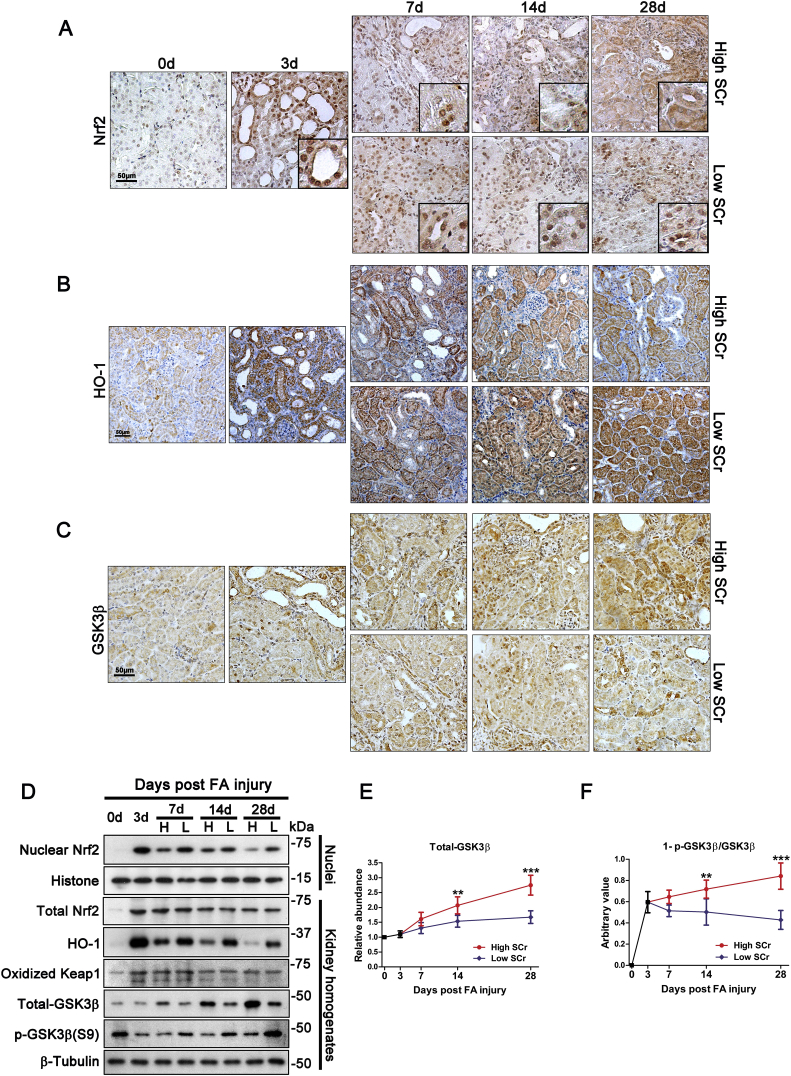
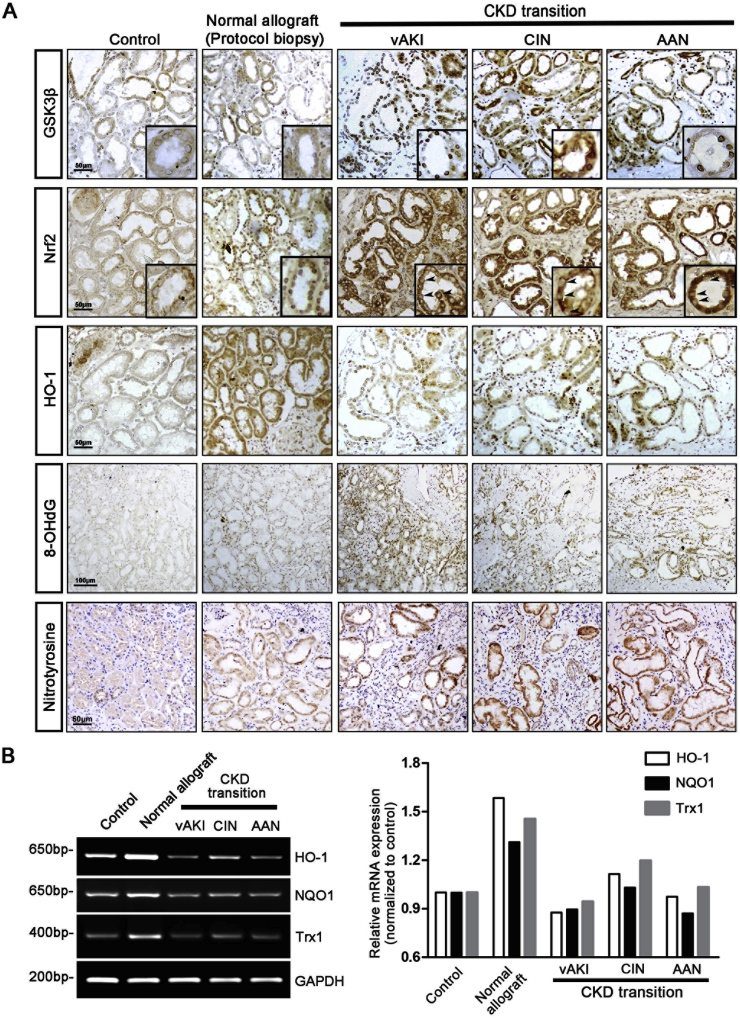
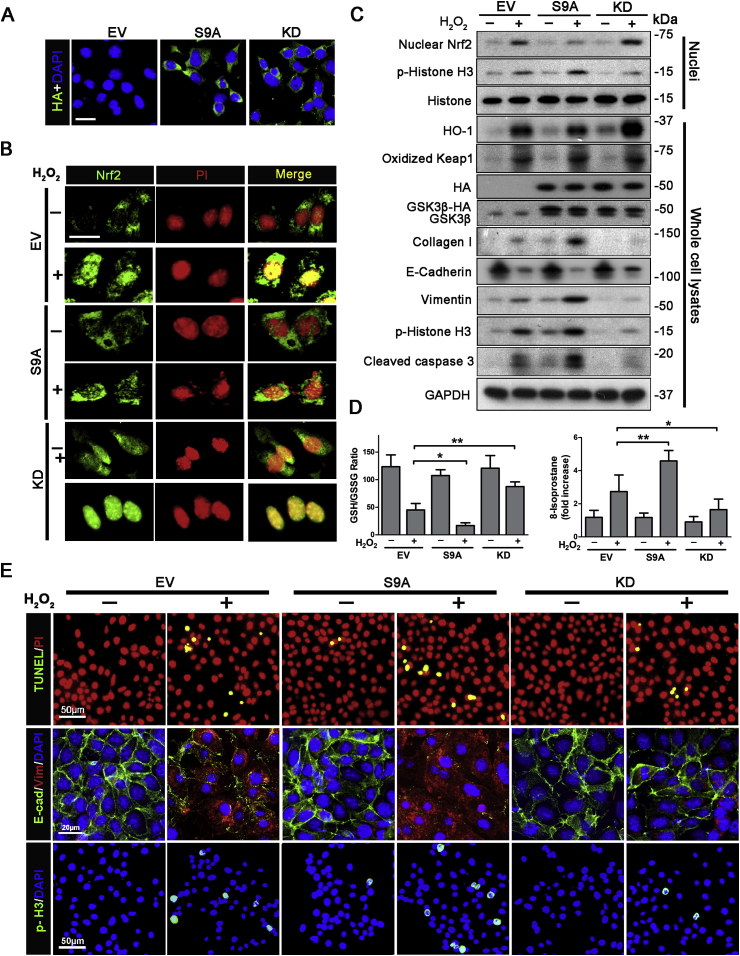
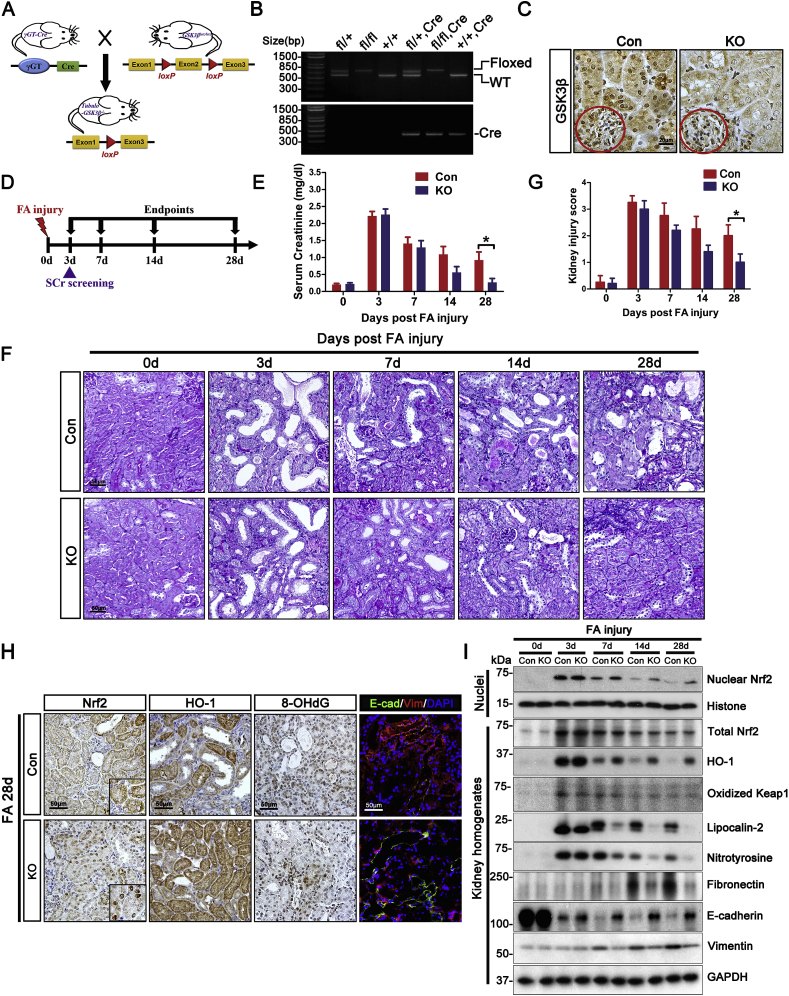
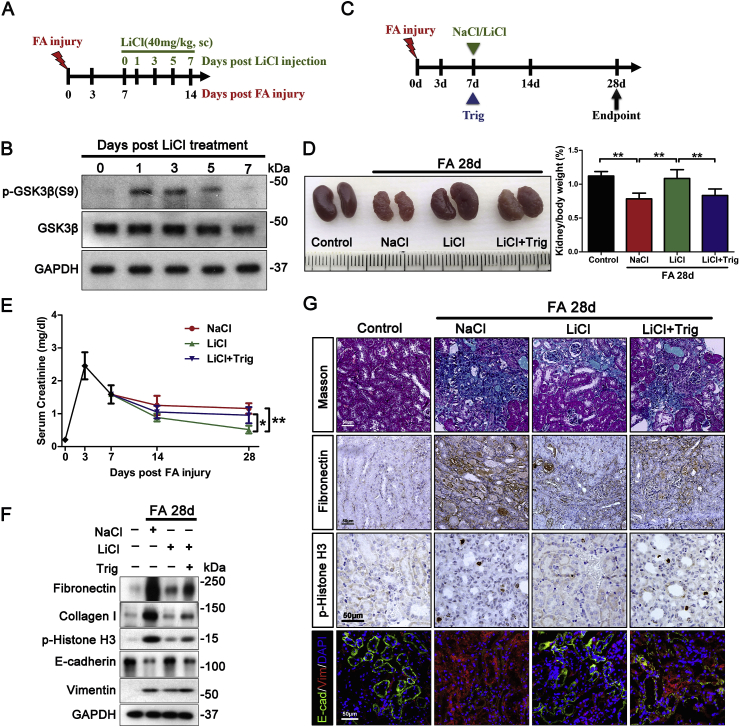
Transition of acute kidney injury (AKI) to chronic kidney disease (CKD) represents an important cause of kidney failure. However, how AKI is transformed into CKD remains elusive. Following folic acid injury, mice developed AKI with ensuing CKD transition, featured by variable degrees of interstitial fibrosis and tubular cell atrophy and growth arrest. This lingering injury of renal tubules was associated with sustained oxidative stress that was concomitant with an impaired Nrf2 antioxidant defense, marked by mitigated Nrf2 nuclear accumulation and blunted induction of its target antioxidant enzymes, like heme oxygenase (HO)-1. Activation of the canonical Keap1/Nrf2 signaling, nevertheless, seems intact during CKD transition because Nrf2 in injured tubules remained activated and elevated in cytoplasm. Moreover, oxidative thiol modification and activation of Keap1, the cytoplasmic repressor of Nrf2, was barely associated with CKD transition. In contrast, glycogen synthase kinase (GSK)3β, a key modulator of the Keap1-independent Nrf2 regulation, was persistently overexpressed and hyperactive in injured tubules. Likewise, in patients who developed CKD following AKI due to diverse etiologies, like volume depletion and exposure to radiocontrast agents or aristolochic acid, sustained GSK3β overexpression was evident in renal tubules and coincided with oxidative damages, impaired Nrf2 nuclear accumulation and mitigated induction of antioxidant gene expression. Mechanistically, the Nrf2 response against oxidative insult was sabotaged in renal tubular cells expressing a constitutively active mutant of GSK3β, but reinforced by ectopic expression of dominant negative GSK3β in a Keap1-independent manner. In vivo in folic acid-injured mice, targeting GSK3β in renal tubules via conditional knockout or by weekly microdose lithium treatment reinstated Nrf2 antioxidant response in the kidney and hindered AKI to CKD transition. Ergo, our findings suggest that GSK3β-mediated Keap1-independent regulation of Nrf2 may serve as an actionable therapeutic target for modifying the long-term sequelae of AKI.
急性肾损伤 (AKI) 向慢性肾脏病 (CKD) 的转变是肾衰竭的一个重要原因。然而,AKI 如何转变为 CKD 仍然难以捉摸。在叶酸损伤后,小鼠发生 AKI,随后发生 CKD 转变,其特征是间质纤维化和肾小管细胞萎缩和生长停滞的程度不同。这种肾小管的持续损伤与持续的氧化应激有关,氧化应激与 Nrf2 抗氧化防御的受损有关,表现在 Nrf2 核积累减少和其靶抗氧化酶如血红素加氧酶 (HO)-1 的诱导减弱。然而,在 CKD 转变过程中,经典的 Keap1/Nrf2 信号的激活似乎是完整的,因为受损的肾小管中的 Nrf2 仍然被激活并在细胞质中升高。此外,氧化硫醇修饰和 Nrf2 的细胞质抑制剂 Keap1 的激活与 CKD 转变几乎没有关联。相比之下,糖原合酶激酶 (GSK)3β,一种 Keap1 非依赖性 Nrf2 调节的关键调节剂,在受损的肾小管中持续过表达和过度活跃。同样,在因各种病因(如容量不足、接触放射性对比剂或马兜铃酸)发生 AKI 后发展为 CKD 的患者中,肾小管中持续存在 GSK3β 过表达,并与氧化损伤、Nrf2 核积累减少和抗氧化基因表达的诱导减弱相一致。从机制上讲,在表达组成性激活的 GSK3β 突变体的肾小管细胞中,Nrf2 对氧化应激的反应被破坏,但通过以 Keap1 非依赖性方式异位表达显性负性 GSK3β 得到加强。在叶酸损伤的小鼠体内,通过条件性敲除或每周微剂量锂治疗靶向肾小管中的 GSK3β,可恢复肾脏中的 Nrf2 抗氧化反应,并阻止 AKI 向 CKD 的转变。因此,我们的研究结果表明,GSK3β 介导的 Nrf2 的 Keap1 非依赖性调节可能成为一种可行的治疗靶点,用于改变 AKI 的长期后果。