Department of Clinical Diagnostic Oncology, Showa University Clinical Research Institute for Clinical Pharmacology and Therapeutics, 6-11-11 kita-karasuyama, setagaya-ku, Tokyo 157-8577, Japan.
Department of Biomedical Sciences, Division of Biochemistry, Nihon University School of Medicine, 30-1 Oyaguchi-kamicho, Itabashi-ku, Tokyo 173-8610, Japan.
Int J Mol Sci. 2019 Jul 29;20(15):3700. doi: 10.3390/ijms20153700.
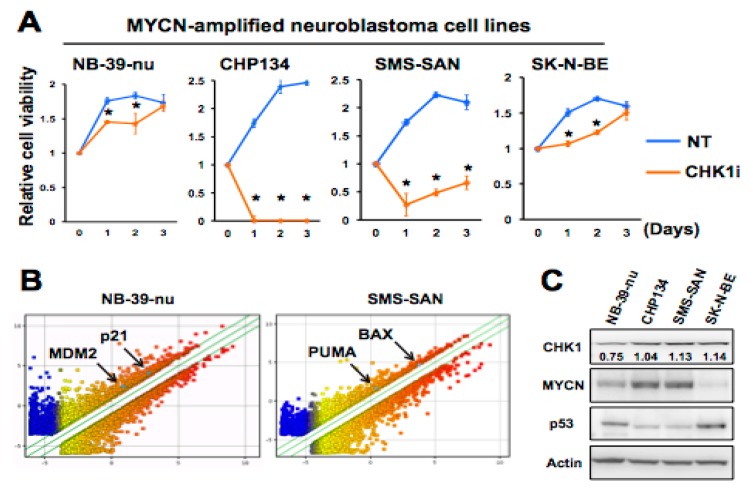
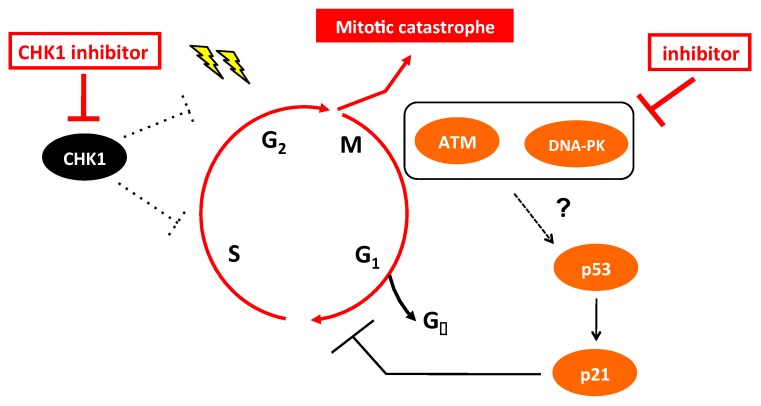
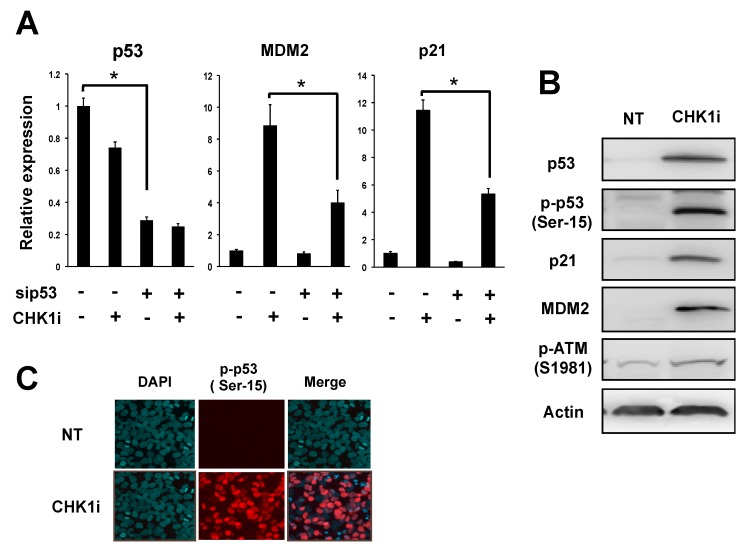
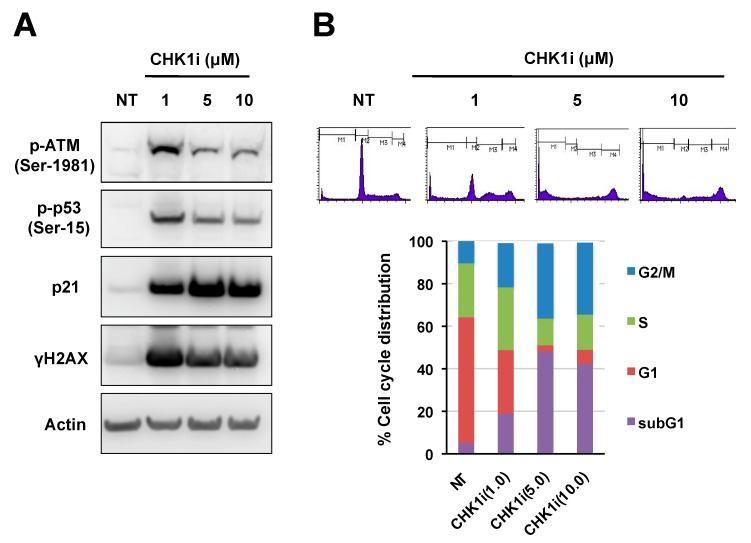
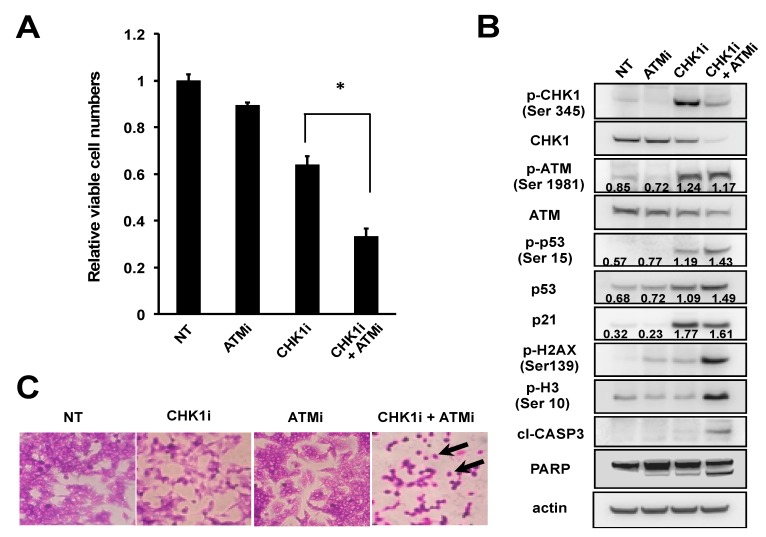
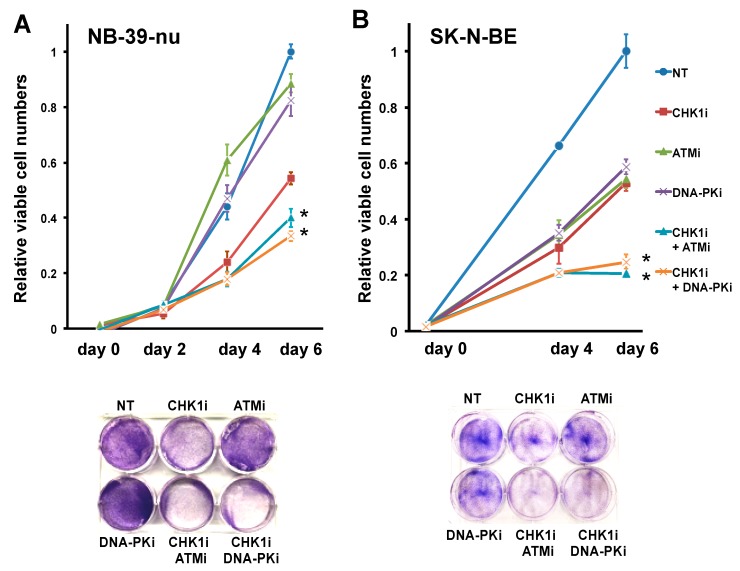
Checkpoint kinase 1 (CHK1) is a central mediator of the DNA damage response (DDR) at the S and G2/M cell cycle checkpoints, and plays a crucial role in preserving genomic integrity. CHK1 overexpression is thought to contribute to cancer aggressiveness, and several selective inhibitors of this kinase are in clinical development for various cancers, including neuroblastoma (NB). Here, we examined the sensitivity of MYCN-amplified NB cell lines to the CHK1 inhibitor PF-477736 and explored mechanisms to increase its efficacy. PF-477736 treatment of two sensitive NB cell lines, SMS-SAN and CHP134, increased the expression of two pro-apoptotic proteins, BAX and PUMA, providing a mechanism for the effect of the CHK1 inhibitor. In contrast, in NB-39-nu and SK-N-BE cell lines, PF-477736 induced DNA double-strand breaks and activated the ataxia telangiectasia mutated serine/threonine kinase (ATM)-p53-p21 axis of the DDR pathway, which rendered the cells relatively insensitive to the antiproliferative effects of the CHK1 inhibitor. Interestingly, combined treatment with PF-477736 and the ATM inhibitor Ku55933 overcame the insensitivity of NB-39-nu and SK-N-BE cells to CHK1 inhibition and induced mitotic cell death. Similarly, co-treatment with PF-477736 and NU7441, a pharmacological inhibitor of DNA-PK, which is also essential for the DDR pathway, rendered the cells sensitive to CHK1 inhibition. Taken together, our results suggest that synthetic lethality between inhibitors of CHK1 and the DDR drives G2/M checkpoint abrogation and could be a novel potential therapeutic strategy for NB.
细胞周期检查点激酶 1(CHK1)是 S 和 G2/M 细胞周期检查点中 DNA 损伤反应(DDR)的中央介质,在维持基因组完整性方面发挥着关键作用。CHK1 的过表达被认为有助于癌症的侵袭性,并且几种该激酶的选择性抑制剂正在针对包括神经母细胞瘤(NB)在内的各种癌症进行临床开发。在这里,我们研究了 MYCN 扩增的 NB 细胞系对 CHK1 抑制剂 PF-477736 的敏感性,并探讨了提高其疗效的机制。PF-477736 处理两种敏感的 NB 细胞系 SMS-SAN 和 CHP134,增加了两种促凋亡蛋白 BAX 和 PUMA 的表达,为 CHK1 抑制剂的作用提供了一种机制。相比之下,在 NB-39-nu 和 SK-N-BE 细胞系中,PF-477736 诱导 DNA 双链断裂并激活 DDR 途径中的共济失调毛细血管扩张突变丝氨酸/苏氨酸激酶(ATM)-p53-p21 轴,这使得细胞对 CHK1 抑制剂的抗增殖作用相对不敏感。有趣的是,PF-477736 与 ATM 抑制剂 Ku55933 的联合治疗克服了 NB-39-nu 和 SK-N-BE 细胞对 CHK1 抑制的不敏感性,并诱导有丝分裂细胞死亡。同样,PF-477736 与 NU7441 的联合治疗(一种也是 DDR 途径所必需的 DNA-PK 的药理学抑制剂)使细胞对 CHK1 抑制敏感。总之,我们的结果表明,CHK1 抑制剂和 DDR 之间的合成致死性驱动 G2/M 检查点缺失,可能成为 NB 的一种新的潜在治疗策略。