Center for Biophysics and Quantitative Biology, University of Illinois Urbana-Champaign, Urbana, IL, 61801, USA.
Department of Computer Science, University of Illinois Urbana-Champaign, Urbana, IL, 61801, USA.
BMC Biol. 2019 Jul 30;17(1):62. doi: 10.1186/s12915-019-0679-8.
Identification of functional non-coding variants and their mechanistic interpretation is a major challenge of modern genomics, especially for precision medicine. Transcription factor (TF) binding profiles and epigenomic landscapes in reference samples allow functional annotation of the genome, but do not provide ready answers regarding the effects of non-coding variants on phenotypes. A promising computational approach is to build models that predict TF-DNA binding from sequence, and use such models to score a variant's impact on TF binding strength. Here, we asked if this mechanistic approach to variant interpretation can be combined with information on genotype-phenotype associations to discover transcription factors regulating phenotypic variation among individuals.
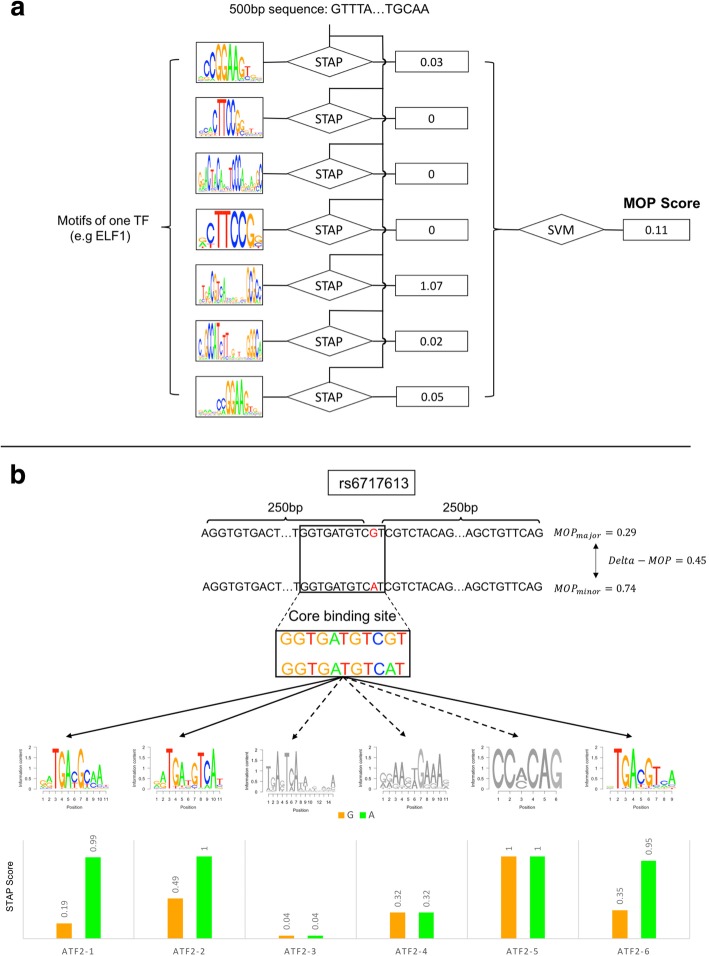
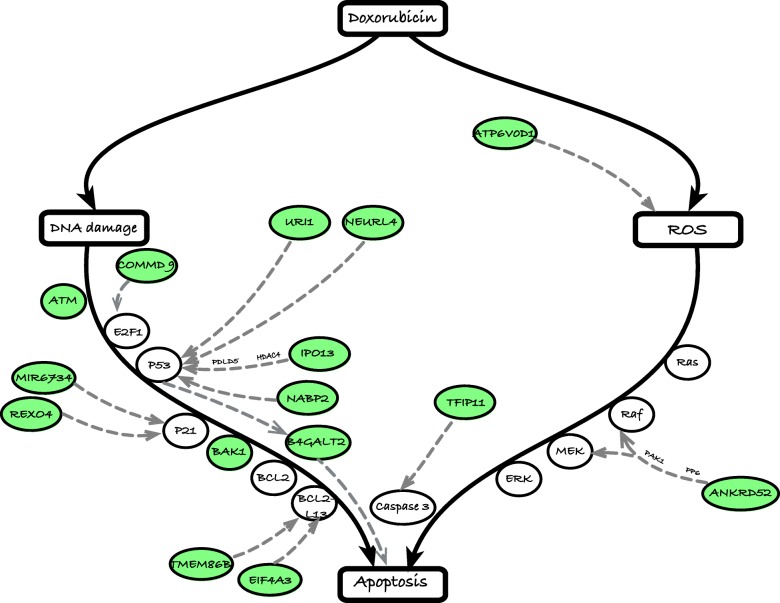
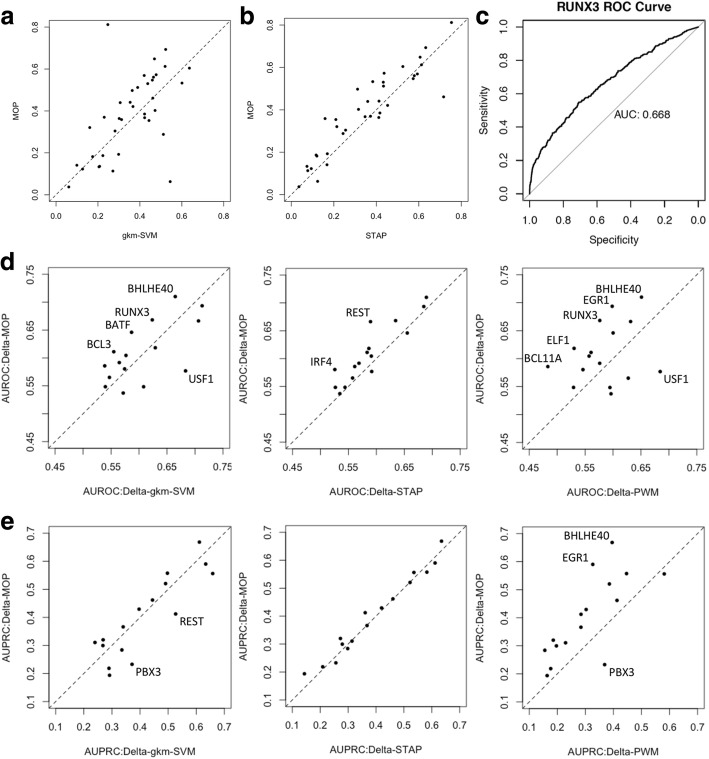
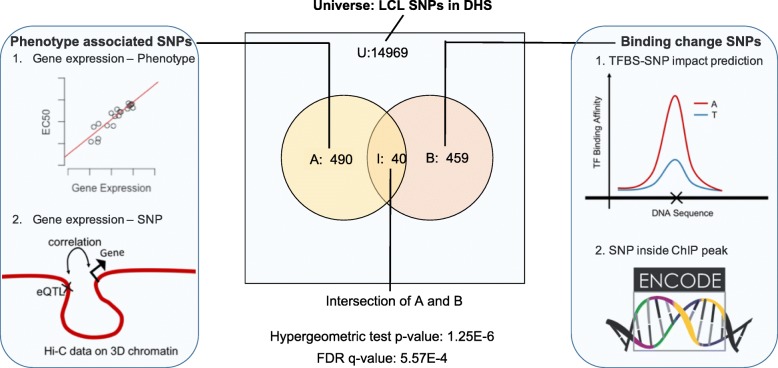
We developed a statistical approach that integrates phenotype, genotype, gene expression, TF ChIP-seq, and Hi-C chromatin interaction data to answer this question. Using drug sensitivity of lymphoblastoid cell lines as the phenotype of interest, we tested if non-coding variants statistically linked to the phenotype are enriched for strong predicted impact on DNA binding strength of a TF and thus identified TFs regulating individual differences in the phenotype. Our approach relies on a new method for predicting variant impact on TF-DNA binding that uses a combination of biophysical modeling and machine learning. We report statistical and literature-based support for many of the TFs discovered here as regulators of drug response variation. We show that the use of mechanistically driven variant impact predictors can identify TF-drug associations that would otherwise be missed. We examined in depth one reported association-that of the transcription factor ELF1 with the drug doxorubicin-and identified several genes that may mediate this regulatory relationship.
Our work represents initial steps in utilizing predictions of variant impact on TF binding sites for discovery of regulatory mechanisms underlying phenotypic variation. Future advances on this topic will be greatly beneficial to the reconstruction of phenotype-associated gene regulatory networks.
鉴定功能非编码变异及其机制解释是现代基因组学的主要挑战,尤其是在精准医学方面。参考样本中的转录因子(TF)结合谱和表观基因组景观可对基因组进行功能注释,但无法针对非编码变异对表型的影响提供明确答案。一种很有前途的计算方法是构建可从序列预测 TF-DNA 结合的模型,并使用此类模型来评估变异对 TF 结合强度的影响。在此,我们探讨了这种用于解释变异的机制方法是否可以与基因型-表型关联信息相结合,从而发现调节个体间表型变异的转录因子。
我们开发了一种统计方法,该方法整合了表型、基因型、基因表达、TF ChIP-seq 和 Hi-C 染色质相互作用数据,以回答这个问题。我们以淋巴母细胞系的药物敏感性作为感兴趣的表型,检验与表型统计上相关的非编码变异是否富集于对 TF 与 DNA 结合强度具有强预测影响的变异,并由此鉴定出调节表型个体差异的 TF。我们的方法依赖于一种新的预测变异对 TF-DNA 结合影响的方法,该方法结合了生物物理建模和机器学习。我们报告了许多在此发现的 TF 作为药物反应变异调节因子的统计和文献支持。我们表明,使用基于机制的变异影响预测因子可以识别否则会被忽略的 TF-药物关联。我们深入研究了一个已报道的关联,即转录因子 ELF1 与药物阿霉素之间的关联,并鉴定出了可能介导这种调控关系的几个基因。
我们的工作代表了利用变异对 TF 结合位点的影响预测来发现表型变异潜在调控机制的初步步骤。在这一主题上的未来进展将极大地有益于与表型相关的基因调控网络的重建。