Department of Biochemistry and Molecular Biology, Virginia Commonwealth University, School of Medicine and the Massey Cancer Center, Richmond, VA, 23298, USA.
Laboratory of Molecular Immunology, National Institutes of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD, 20892, USA.
J Neuroinflammation. 2019 Jul 30;16(1):161. doi: 10.1186/s12974-019-1548-7.
Multiple sclerosis (MS) is an autoimmune demyelinating disease of the central nervous system (CNS). It is firmly established that overactivation of the p65 (RelA) nuclear factor kappa B (NF-κB) transcription factor upregulates expression of inflammatory mediators in both immune and non-immune resident CNS cells and promotes inflammation during MS. In contrast to p65, NF-κB family member RelB regulates immune cell development and can limit inflammation. Although RelB expression is induced during inflammation in the CNS, its role in MS remains unknown.
To examine the role of RelB in non-immune CNS cells, we generated mice with RelB specifically deleted in astrocytes (RelB), oligodendrocytes (RelB), or neural progenitor-derived cells (RelB). We used experimental autoimmune encephalomyelitis (EAE), an accepted mouse model of MS, to assess the effect of RelB deletion on disease outcomes and performed analysis on the histological, cellular, and molecular level.
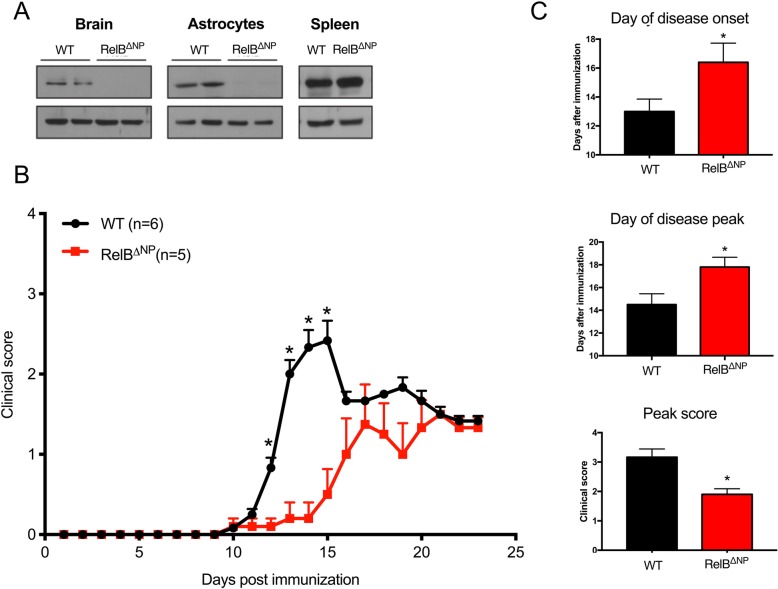
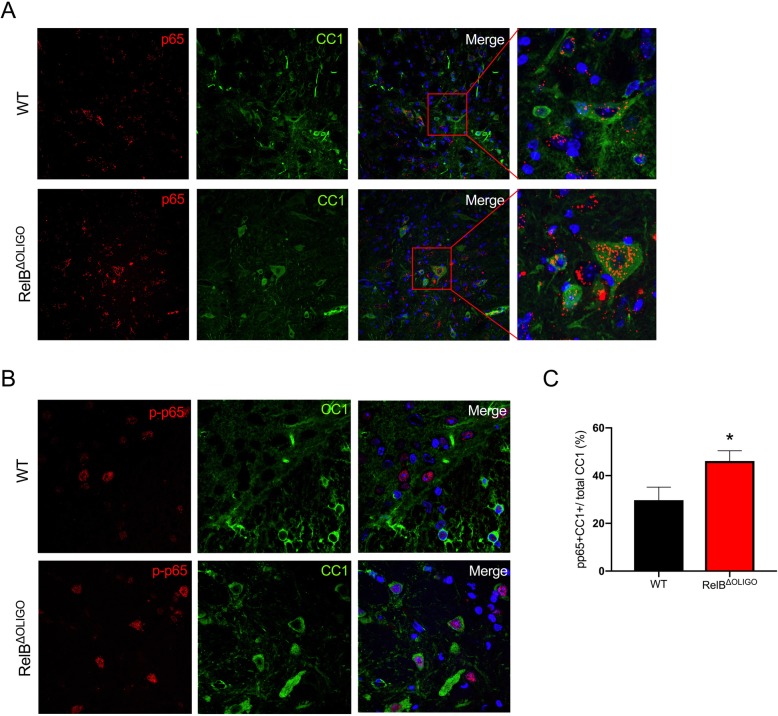
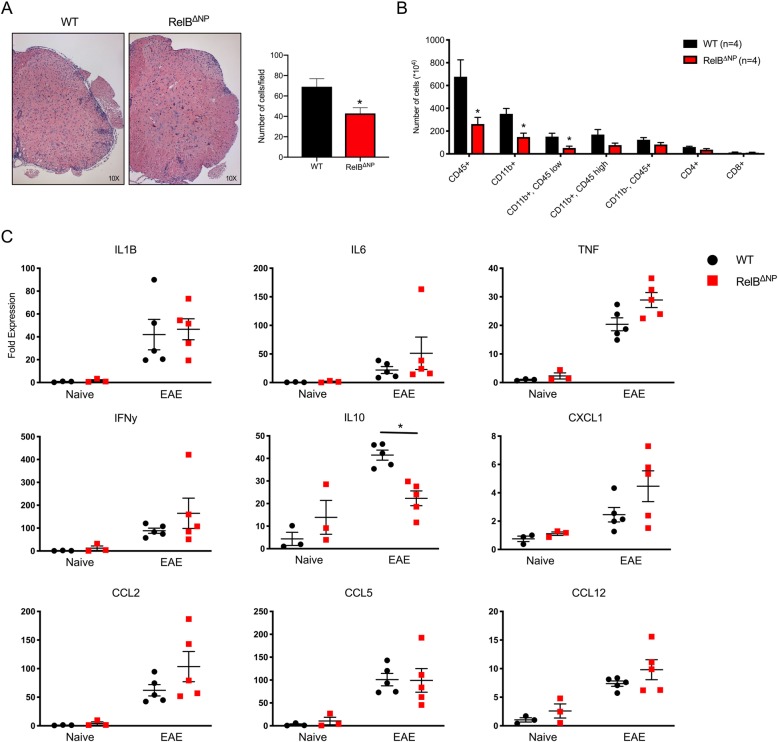
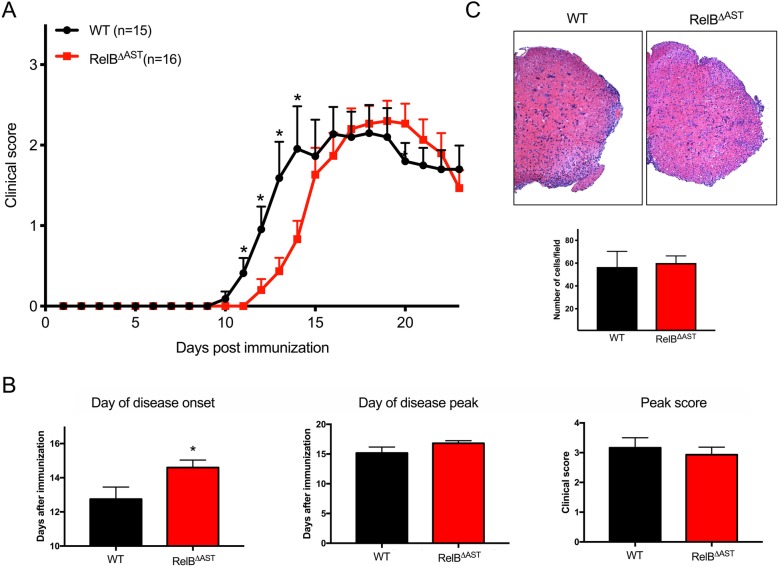
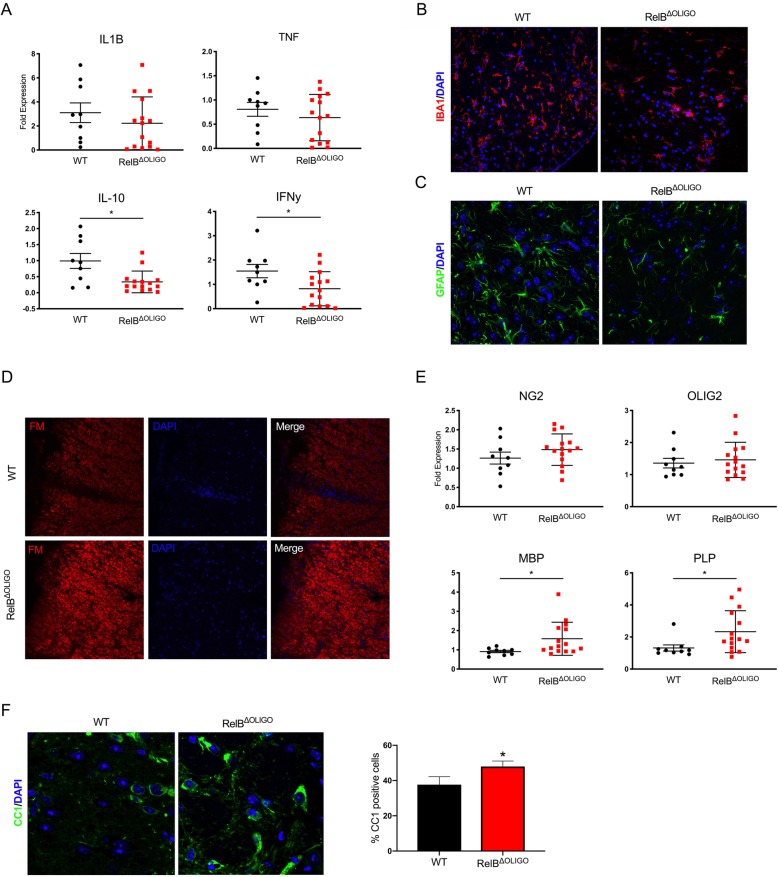
Despite being a negative regulator of inflammation, conditional knockout of RelB in non-immune resident CNS cells surprisingly decreased the severity of EAE. This protective effect was recapitulated by conditional deletion of RelB in oligodendrocytes but not astrocytes. Deletion of RelB in oligodendrocytes reduced disease severity, promoted survival of mature oligodendrocytes, and correlated with increased activation of p65 NF-κB.
These findings suggest that RelB fine tunes inflammation and cell death/survival during EAE. Importantly, our data points out the detrimental role RelB plays in controlling survival of mature oligodendrocytes, which could be explored as a viable option to treat MS in the future.
多发性硬化症(MS)是一种中枢神经系统(CNS)自身免疫性脱髓鞘疾病。现已明确,p65(RelA)核因子κB(NF-κB)转录因子的过度激活会上调免疫和非免疫中枢神经系统固有细胞中炎症介质的表达,并在 MS 期间促进炎症。与 p65 不同,NF-κB 家族成员 RelB 调节免疫细胞的发育,并能限制炎症。尽管 RelB 在中枢神经系统炎症过程中被诱导表达,但它在 MS 中的作用尚不清楚。
为了研究 RelB 在非免疫中枢神经系统细胞中的作用,我们生成了特异性在星形胶质细胞(RelB)、少突胶质细胞(RelB)或神经祖细胞衍生细胞(RelB)中缺失 RelB 的小鼠。我们使用实验性自身免疫性脑脊髓炎(EAE),一种公认的 MS 小鼠模型,评估 RelB 缺失对疾病结果的影响,并在组织学、细胞和分子水平上进行分析。
尽管 RelB 是炎症的负调节剂,但在非免疫中枢神经系统固有细胞中条件性敲除 RelB 出人意料地降低了 EAE 的严重程度。这种保护作用在少突胶质细胞中条件性敲除 RelB 时重现,但在星形胶质细胞中则不然。在少突胶质细胞中敲除 RelB 可减轻疾病严重程度,促进成熟少突胶质细胞的存活,并与 p65 NF-κB 的激活增加相关。
这些发现表明,RelB 在 EAE 期间精细地调节炎症和细胞死亡/存活。重要的是,我们的数据表明,RelB 在控制成熟少突胶质细胞的存活方面起着有害作用,这可能成为未来治疗 MS 的可行选择。