Vujosevic Snezana, Medenica Sanja, Vujicic Vesko, Dapcevic Milena, Bakic Nikola, Yang Ruhua, Liu Jun, Mistry Pramod K
Department of Endocrinology, Internal Medicine Clinic, Clinical Center of Montenegro, Faculty of Medicine, University of Montenegro, Podgorica 81000, Montenegro.
Hematology Department, Internal Medicine Clinic, Clinical Center of Montenegro, Faculty of Medicine, University of Montenegro, Podgorica 81000, Montenegro.
World J Clin Cases. 2019 Jun 26;7(12):1475-1482. doi: 10.12998/wjcc.v7.i12.1475.
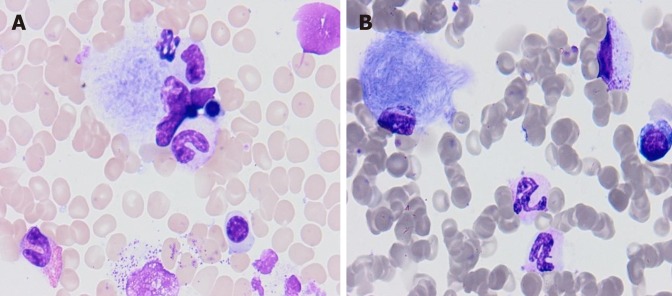
The most common lysosomal storage disorder is Gaucher disease (GD). It is a deficiency of lysosomal glucocerebrosidase (GBA) due to biallelic mutations in the gene, characterized by the deposition of glucocerebroside in macrophage-monocyte system cells. The report targets clinical phenotypes of GD in order to correlate them with gene mutations, as well as to identify gene mutation in patients in Montenegro that are diagnosed with GD.
Five patients (4 male, 1 female) of type 1 GD (GD1) are reported. The age at diagnosis ranged from 7 to 40. Patients experienced delays of 1-12 years in diagnosis after the original onset of symptoms. The most common mode of presentation was a variable degree of splenomegaly and thrombocytopenia, while other symptoms included bone pain, hepatomegaly, abdominal pain and fatigue. Osteopenia was present in a majority of the patients: 4/5. All patients were found to have an asymptomatic Erlenmeyer flask deformity of the distal femur. On enzyme replacement therapy (ERT), the hematological and visceral parameters showed significant improvement, but no significant progression in bone mineral density was noticed. gene sequencing revealed homozygosity for the mutation in one patient. The genotypes of the other patients were N370S/55bp deletion, N370S/D409H (2 patients), and H255Q/N370S (1 patient).
The phenotypes of the GD1 encountered in Montenegro were severe but all responded well to ERT.
最常见的溶酶体贮积症是戈谢病(GD)。它是由于该基因的双等位基因突变导致溶酶体葡萄糖脑苷脂酶(GBA)缺乏,其特征是葡萄糖脑苷脂在巨噬细胞 - 单核细胞系统细胞中沉积。本报告旨在研究GD的临床表型,以便将它们与基因突变相关联,同时鉴定黑山地区被诊断为GD的患者的基因突变情况。
报告了5例1型戈谢病(GD1)患者(4例男性,1例女性)。诊断时的年龄在7至40岁之间。患者在最初出现症状后1 - 12年才得以诊断。最常见的表现形式是不同程度的脾肿大和血小板减少,其他症状包括骨痛、肝肿大、腹痛和疲劳。大多数患者(4/5)存在骨质减少。所有患者均被发现有股骨远端无症状的烧瓶样畸形。接受酶替代疗法(ERT)后,血液学和内脏参数显示出显著改善,但骨密度未发现明显进展。基因测序显示1例患者存在该突变的纯合性。其他患者的基因型分别为N370S/55bp缺失、N370S/D409H(2例患者)和H255Q/N370S(1例患者)。
黑山地区遇到的GD1表型严重,但所有患者对ERT反应良好。