Ou Li, Kim Sarah, Whitley Chester B, Jarnes-Utz Jeanine R
Gene Therapy Center, Department of Pediatrics, University of Minnesota, United States of America.
Department of Experimental and Clinical Pharmacology, College of Pharmacy, University of Minnesota, United States of America.
Mol Genet Metab Rep. 2019 Jul 17;20:100495. doi: 10.1016/j.ymgmr.2019.100495. eCollection 2019 Sep.
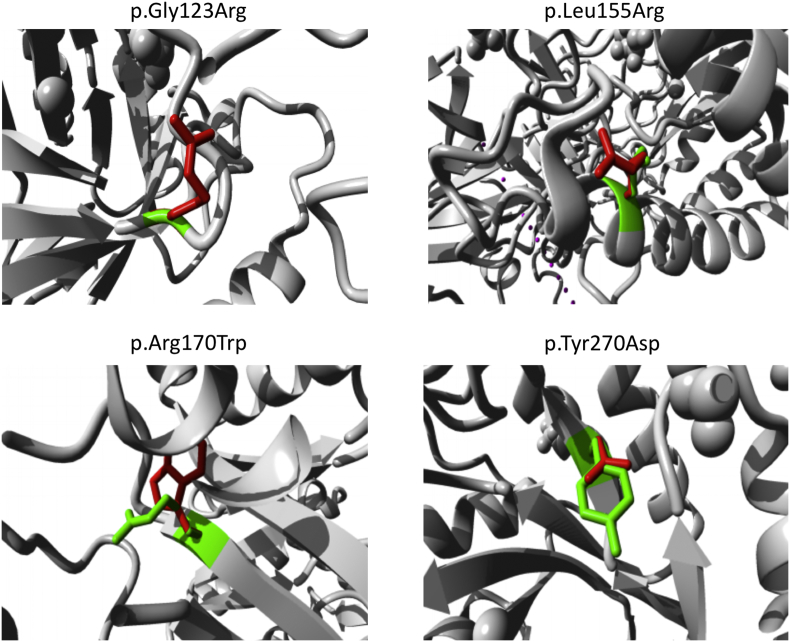
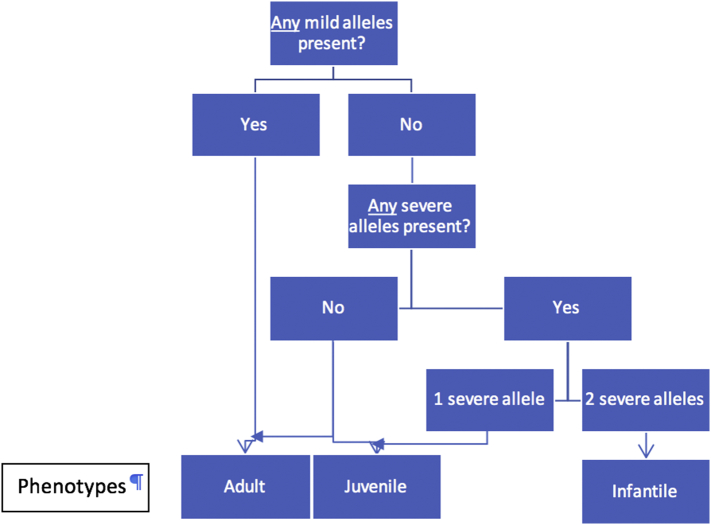
Gangliosidoses, including GM1-gangliosidosis and GM2-gangliosidosis (Tay-Sachs disease and Sandhoff disease), are lysosomal disorders resulting from enzyme deficiencies and accumulation of gangliosides. Phenotypes of gangliosidoses range from infantile, late-infantile, juvenile, and to the adult form. The genotype-phenotype correlation is essential for prognosis and clinical care planning for patients with a gangliosidosis condition. Previously, we have developed a method to establish the genotype-phenotype correlation of another lysosomal disease, mucopolysaccharidosis type I, with in silico tools. This same method was applied to analyze the genotype and phenotype of 38 patients diagnosed with a gangliosidosis disease in the United States. Out of 40 mutations identified, 3 were novel, including p.Tyr192His and p.Phe556Ser of the gene and p.Gly461Val of the gene. Furthermore, the mutant protein structure of all missense mutations was constructed by homology modeling. A systemic structural analysis of these models revealed the specific mechanisms of how each mutation may lead to the disease. In summary, the method developed in this study holds promise as a tool that can be broadly applicable to other lysosomal diseases and monogenic diseases.
神经节苷脂贮积症,包括GM1神经节苷脂贮积症和GM2神经节苷脂贮积症(泰-萨克斯病和桑德霍夫病),是由于酶缺乏和神经节苷脂蓄积导致的溶酶体疾病。神经节苷脂贮积症的表型范围从婴儿型、晚婴儿型、青少年型到成人型。基因型-表型相关性对于神经节苷脂贮积症患者的预后和临床护理规划至关重要。此前,我们已经开发出一种方法,利用计算机工具建立另一种溶酶体疾病——I型黏多糖贮积症的基因型-表型相关性。同样的方法被用于分析在美国诊断为神经节苷脂贮积症的38例患者的基因型和表型。在鉴定出的40个突变中,有3个是新的,包括 基因的p.Tyr192His和p.Phe556Ser以及 基因的p.Gly461Val。此外,通过同源建模构建了所有错义突变的突变蛋白结构。对这些模型的系统结构分析揭示了每个突变可能导致疾病的具体机制。总之,本研究开发的方法有望成为一种可广泛应用于其他溶酶体疾病和单基因疾病的工具。