Muangnoi Chawanphat, Ratnatilaka Na Bhuket Pahweenvaj, Jithavech Ponsiree, Wichitnithad Wisut, Srikun Onsiri, Nerungsi Chakkrapan, Patumraj Suthiluk, Rojsitthisak Pornchai
Pharmaceutical Chemistry and Natural Products Program, Faculty of Pharmaceutical Sciences, Chulalongkorn University, Bangkok 10330, Thailand.
Natural Products for Ageing and Chronic Diseases Research Unit, Chulalongkorn University, Bangkok 10330, Thailand.
Pharmaceutics. 2019 Aug 1;11(8):373. doi: 10.3390/pharmaceutics11080373.
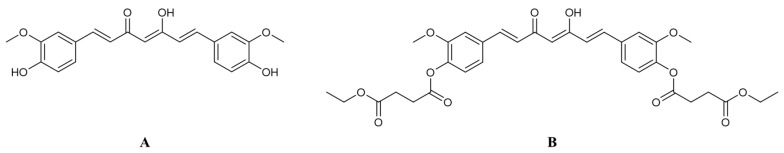
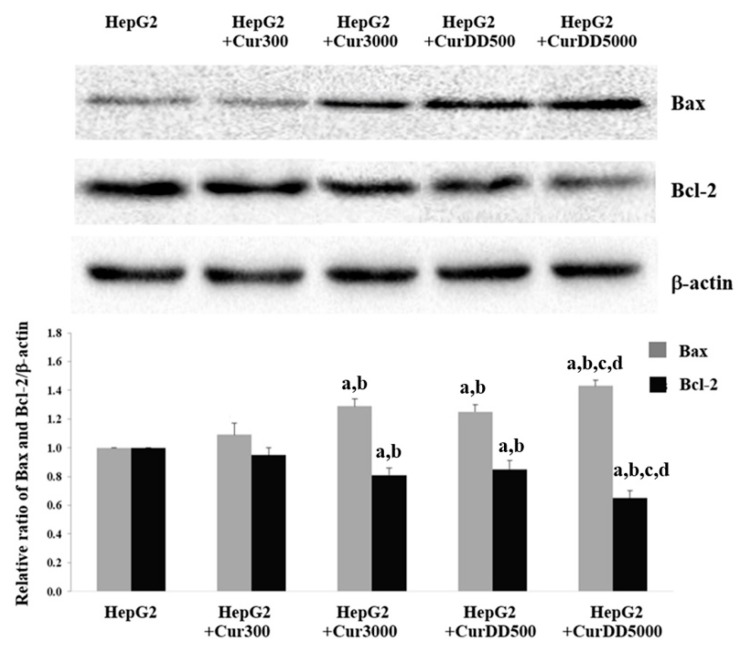
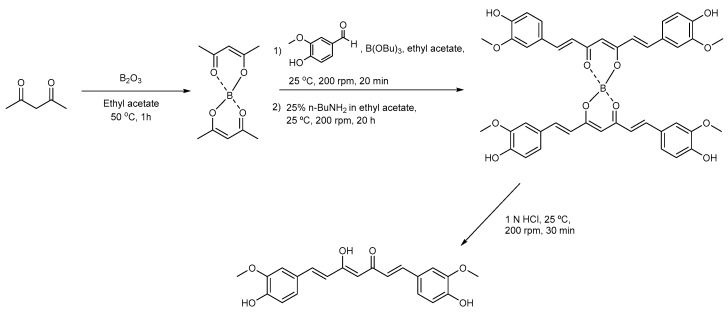
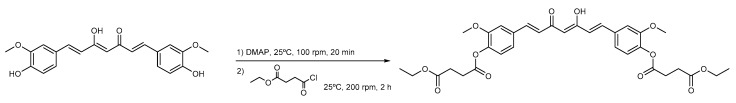
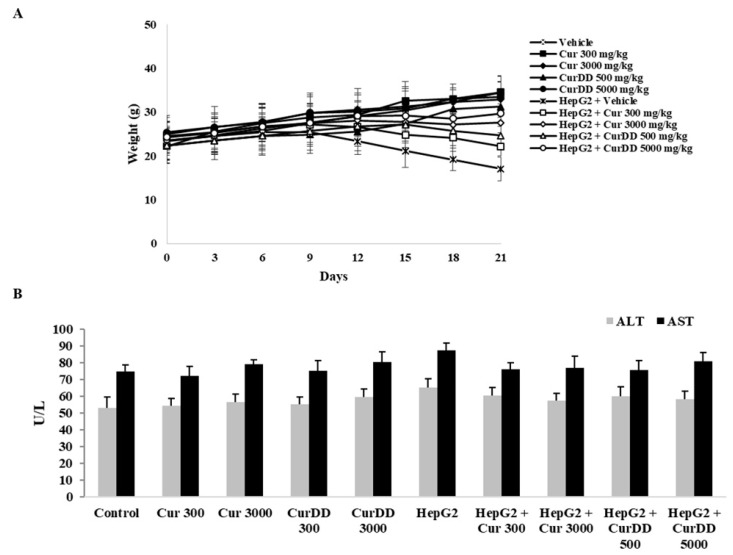
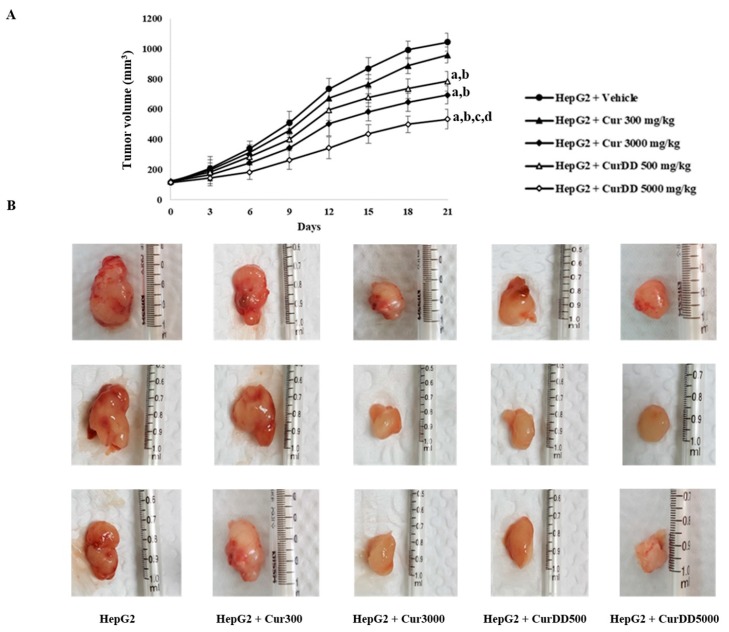
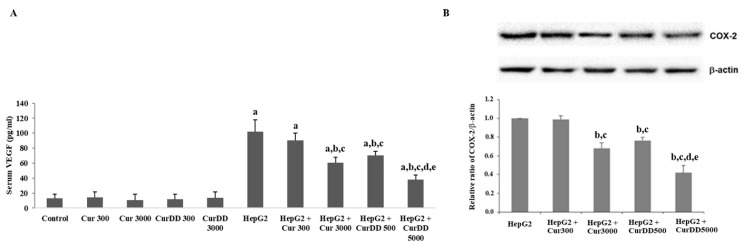
Previously, we synthesized curcumin and a succinate ester prodrug of curcumin namely curcumin diethyl disuccinate (CurDD) in the lab scale, which yielded hundred milligrams to few grams of the compounds. CurDD was found to be more stable in a phosphate buffer pH 7.4 and exhibited better cytotoxicity against Caco-2 cells than curcumin. Here, the one-pot syntheses of curcumin and CurDD were scaled up to afford multigram quantities of both compounds for preclinical studies using a 10-L chemical reactor. The key steps for the synthesis of curcumin were the formation of boron-acetylacetone complex and the decomplexation of boron-curcumin complex. The synthesis of CurDD could be achieved via a one-step esterification between curcumin and succinic acid monoethyl ester chloride using 4-(,-dimethylamino)pyridine as a catalyst. The synthesized curcumin and CurDD were then investigated and compared for an anti-tumor activity in HepG2-xenograft mice. CurDD could reduce the tumor growth in HepG2-xenograft mice better than curcumin. CurDD also exerted the stronger inhibition on VEGF secretion, COX-2 and Bcl-2 expression and induced higher Bax expression in comparison with curcumin. The results suggest that CurDD is a promising prodrug of curcumin and has a potential to be further developed as a therapeutic agent or an adjuvant for the treatment of hepatocellular carcinoma.
此前,我们在实验室规模下合成了姜黄素及其琥珀酸酯前药姜黄素二乙基二琥珀酸酯(CurDD),产量为数百毫克至几克该化合物。发现CurDD在pH 7.4的磷酸盐缓冲液中更稳定,并且对Caco-2细胞的细胞毒性比姜黄素更好。在此,使用10升化学反应器将姜黄素和CurDD的一锅法合成放大,以提供多克量的这两种化合物用于临床前研究。姜黄素合成的关键步骤是硼 - 乙酰丙酮络合物的形成和硼 - 姜黄素络合物的解络合。CurDD的合成可以通过使用4 -(N,N - 二甲基氨基)吡啶作为催化剂,在姜黄素和琥珀酸单乙酯氯之间进行一步酯化来实现。然后研究并比较了合成的姜黄素和CurDD在HepG2异种移植小鼠中的抗肿瘤活性。CurDD比姜黄素能更好地抑制HepG2异种移植小鼠中的肿瘤生长。与姜黄素相比,CurDD对VEGF分泌、COX - 2和Bcl - 2表达的抑制作用更强,并诱导更高的Bax表达。结果表明,CurDD是一种有前景的姜黄素前药,有潜力进一步开发成为治疗肝细胞癌的治疗剂或佐剂。