Standage Daniel S, Brown C Titus, Hormozdiari Fereydoun
Population Health and Reproduction, University of California, Davis, USA.
Population Health and Reproduction, University of California, Davis, USA; Genome Center, University of California, Davis, USA.
iScience. 2019 Aug 30;18:28-36. doi: 10.1016/j.isci.2019.07.032. Epub 2019 Jul 23.
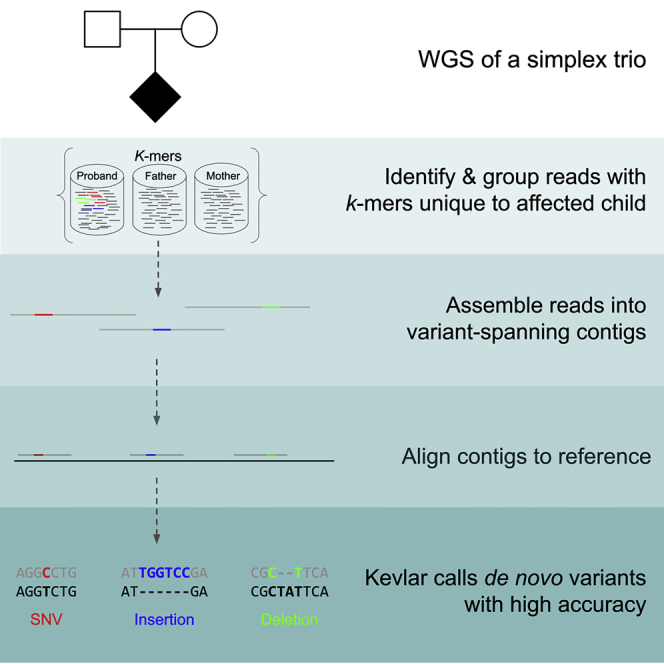
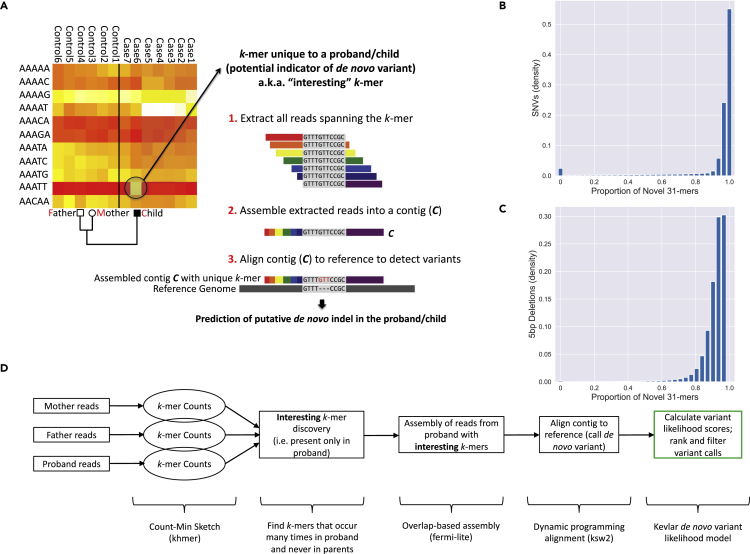
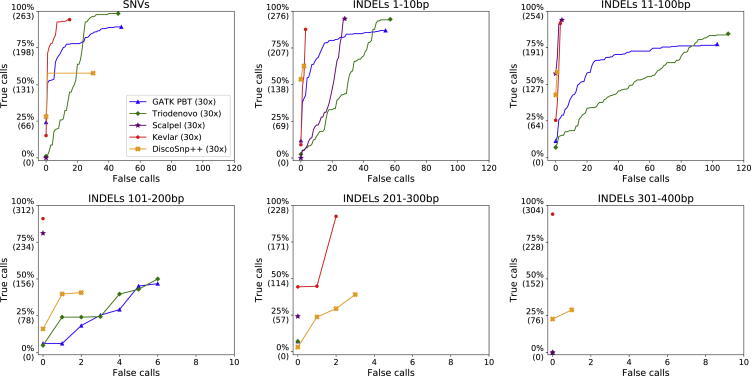
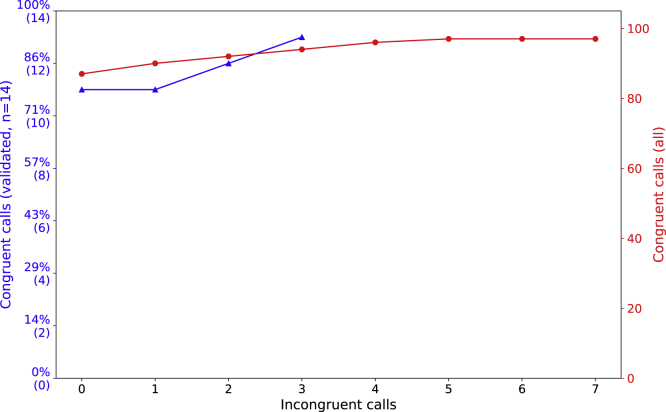
De novo genetic variants are an important source of causative variation in complex genetic disorders. Many methods for variant discovery rely on mapping reads to a reference genome, detecting numerous inherited variants irrelevant to the phenotype of interest. To distinguish between inherited and de novo variation, sequencing of families (parents and siblings) is commonly pursued. However, standard mapping-based approaches tend to have a high false-discovery rate for de novo variant prediction. Kevlar is a mapping-free method for de novo variant discovery, based on direct comparison of sequences between related individuals. Kevlar identifies high-abundance k-mers unique to the individual of interest. Reads containing these k-mers are partitioned into disjoint sets by shared k-mer content for variant calling, and preliminary variant predictions are sorted using a probabilistic score. We evaluated Kevlar on simulated and real datasets, demonstrating its ability to detect both de novo single-nucleotide variants and indels with high accuracy.
新生基因变异是复杂遗传疾病中致病变异的重要来源。许多变异发现方法依赖于将 reads 映射到参考基因组,从而检测出许多与感兴趣的表型无关的遗传变异。为了区分遗传变异和新生变异,通常会对家系(父母和兄弟姐妹)进行测序。然而,基于标准映射的方法在新生变异预测方面往往具有较高的假发现率。Kevlar 是一种用于新生变异发现的无映射方法,基于对相关个体之间序列的直接比较。Kevlar 识别出感兴趣个体特有的高丰度 k-mer。包含这些 k-mer 的 reads 通过共享的 k-mer 内容被划分为不相交的集合用于变异调用,并且使用概率分数对初步的变异预测进行排序。我们在模拟和真实数据集上评估了 Kevlar,证明了它能够高精度地检测新生单核苷酸变异和插入缺失。